商业广告QQ
896000434
896000434
(AD)的发病机制十分复杂,之前的全基因组关联研究发现了多个与先天通路调节相关的AD风险基因[1-3]。小胶质细胞是大脑中的先天免疫细胞,能够通过促炎和吞噬功能促进神经退行性病变。
小胶质细胞可以分为稳态(M0)和神经退行性小胶质细胞(MGnD)两个类型,但对于MGnD的功能和在AD发病机制中发挥的作用还不是很清楚。
在上周的《自然 神经科学》杂志上,哈佛大学医学院和波士顿大学医学院的研究人员发表了最新研究成果[4],他们发现,小胶质细胞的microRNA-155(miR-155)缺失可以通过干扰素- (IFN- )信号诱导前MGnD响应,并且增强吞噬功能和对 淀粉样蛋白(A )斑块的压缩,减少营养不良性神经突,增强对突触的保护,改善认知功能。

研究团队此前已经发现过,在MGnD中,miR-155可以被TREM2-APOE信号通路激活[5],miR-155的全身缺失会加速小鼠A 的沉积[6],而小胶质细胞特异性缺失则会增加溶酶体中A 1-42原纤维[7],表明miR-155可以调节A 的降解。
在这次的研究中,研究人员首先确定了小胶质细胞miR-155的功能,研究人员对AD模式APP/PS1小鼠小胶质细胞的miR-155进行了条件性敲除(cKO)。
8月龄miR-155 cKO小鼠相比野生型小鼠,M0稳态基因表达显著减少,雌性尤为明显,而相比4月龄APP/PS1小鼠,MGnD基因表达显著增加,同时M0基因表达被抑制。APP/PS1小鼠相比野生型小鼠表达增加最多的20个MGnD基因在miR-155 cKO小鼠中进一步大量增加。
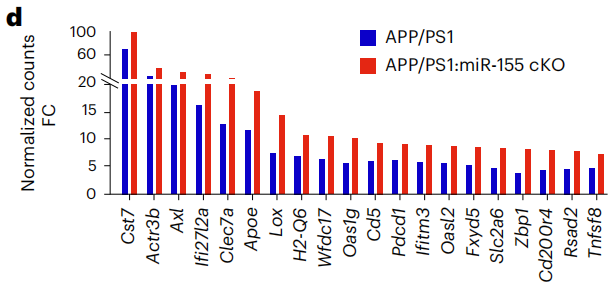
20个MGnD基因在miR-155 cKO小鼠中表达增加更多
APP/PS1与miR-155 cKO小胶质细胞间共享的差异化表达基因与IFN通路增强有关,表明IFN通路是小胶质细胞对远期病理变化的早期响应。miR-155 cKO小胶质细胞表达增加的基因与吞噬作用、抗原呈递和细胞对IFN- 的响应有关。
利用单细胞测序计数,研究人员从4月龄APP/PS1与miR-155 cKO小鼠大脑中分离出了M0、前MGnD和MGnD三类细胞,他们注意到,相比APP/PS1小鼠,miR-155 cKO小鼠的前MGnD规模更大,前MGnD的特征基因表达也明显更高。
前MGnD中的早期和晚期IFN- 响应性前MGnD表现出Stat1、Isg15、Ifi204、Irf7、Ifit1和Ifit3基因表达增加,与之前发现的IFN响应性小胶质细胞非常相似。早期前MGnD保留了M0基因的高表达和MGnD基因的低表达,晚期前MGnD则更多的表达MGnD基因,降低了M0基因的表达。
伪时间分析确认,通过IFN- 响应,小胶质细胞可以从稳态转换到MGnD,而miR-155敲除增加了前MGnD状态的规模和免疫响应。
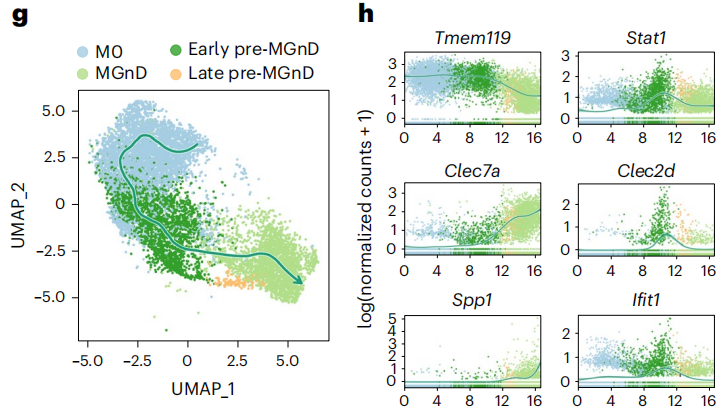
小胶质细胞由M0向MGnD的转换的轨迹
这种转变对AD病理来说,会产生怎样的影响呢?
研究人员发现,4月龄miR-155 cKO小鼠大脑中A 斑块数量和面积显著减少,但8月龄的却没有这种变化。在4月龄miR-155 cKO小鼠中,MGnD对A 的吞噬作用显著增强,这可能意味着,miR-155 cKO MGnD对疾病早期的A 斑块更敏感。
不但如此,miR-155 cKO MGnD还改变了A 斑块的形态。此前有研究[8]发现,小胶质细胞可以紧紧裹住早期形成的淀粉样纤维和斑块,压缩并且让它们与外界 绝缘 ,而存在功能障碍的小胶质细胞包裹能力下降,斑块变得松散,触碰到了更多相邻的神经突,将自身毒性扩散了过去,导致了周围也发生神经炎性tau蛋白过度磷酸化和营养不良性神经突。
然而!miR-155 cKO小胶质细胞重新恢复了压缩和绝缘功能,限制了A 的毒性扩散。
这种保护作用恢复的外在表现体现在,miR-155 cKO小鼠大脑皮层中斑块周围区域的突触数量明显增加,8月龄miR-155 cKO小鼠的认知功能改善,在Y迷宫实验中,自发交替任务相比普通APP/PS1小鼠完成得更好,对迷宫中未探索过的新臂探索增加,意味着功能性识别记忆能力的恢复。
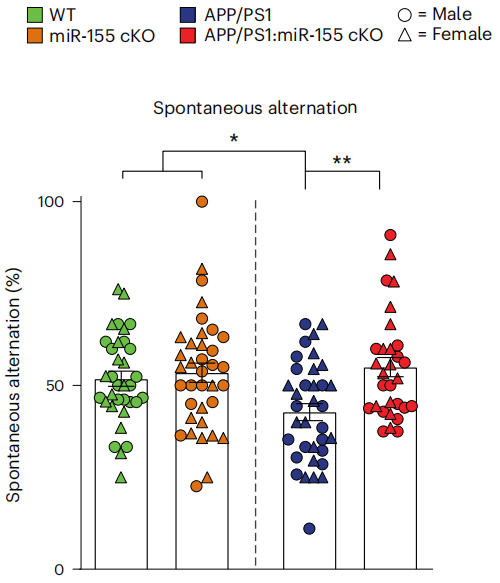
8月龄miR-155 cKO小鼠(红)的表现显著强于同龄APP/PS1小鼠(蓝)
总的来说,这项研究阐明了miR-155介导的MGnD调节机制,以及IFN- 响应性前MGnD对AD小鼠神经退行性病理和保持认知功能的有益作用,突出了miR-155和IFN- 作为AD潜在治疗靶点的可能性。
参考文献:
[1] Efthymiou A G, Goate A M. Late onset Alzheimer s disease genetics implicates microglial pathways in disease risk[J]. Molecular neurodegeneration, 2017, 12(1): 1-12.
[2] Lambert J C, Ibrahim-Verbaas C A, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer s disease[J]. Nature genetics, 2013, 45(12): 1452-1458.
[3] Kunkle B W, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer s disease identifies new risk loci and implicates A , tau, immunity and lipid processing[J]. Nature genetics, 2019, 51(3): 414-430.
[4] Yin, Z., Herron, S., Silveira, S. et al. Identification of a protective microglial state mediated by miR-155 and interferon- signaling in a mouse model of Alzheimer s disease. Nat Neurosci (2023). https://doi.org/10.1038/s41593-023-01355-y
[5] Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases[J]. Immunity, 2017, 47(3): 566-581. e9.
[6] Readhead B, Haure-Mirande J V, Mastroeni D, et al. miR155 regulation of behavior, neuropathology, and cortical transcriptomics in Alzheimer s disease[J]. Acta neuropathologica, 2020, 140: 295-315.
[7] Aloi M S, Prater K E, Sopher B, et al. The pro‐inflammatory microRNA miR‐155 influences fibrillar ‐Amyloid1‐42 catabolism by microglia[J]. Glia, 2021, 69(7): 1736-1748.
[8] Yuan P, Condello C, Keene C D, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy[J]. Neuron, 2016, 90(4): 724-739.
版权声明 本网站所有注明“来源:100医药网”或“来源:bioon”的文字、图片和音视频资料,版权均属于100医药网网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:100医药网”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 87%用户都在用100医药网APP 随时阅读、评论、分享交流 请扫描二维码下载->