商业广告QQ
896000434
896000434
来源:100医药网 2024-03-31 10:35
本研究揭示了一种新的机制,即C5a-C5aR1轴上调Drp1S616的磷酸化促进线粒体分裂,从而促进LN的足细胞损伤。系统性红斑(SLE)是一种以耐受丧失为特征的全身性自身免疫性疾病,导致自身抗体的产生和器官损害。狼疮性肾炎(LN)是系统性红斑狼疮(SLE)患者最常见、最严重的临床表现之一。尽管近几十年来LN的预后有所改善,但5%-20%的LN患者在10年内进展为终末期肾病(ESRD),这突显了寻找新的有效治疗LN靶点的迫切需要。

图片来源:
近日,来自中山大学的研究者们在Molecular Therapy杂志上发表了题为 C5a-C5aR1 axis controls mitochondrial fission to promote podocyte injury in lupus nephritis 的文章,该研究揭示了C5a-C5aR1轴控制线粒体分裂促进狼疮性肾炎足细胞损伤。
足细胞对维持肾小球滤过屏障的完整性是必不可少的,但在狼疮性肾炎(LN)中它们经常受到影响。在本研究中,研究者表明,足细胞中Drp1S616磷酸化的显著上调促进了线粒体的分裂,导致了LN的线粒体和足细胞损伤。
抑制或敲除Drp1可促进线粒体融合,保护足细胞免受LN血清的损伤。在体内,对Drp1的药理抑制减少了狼疮易感小鼠足细胞中Drp1S616的磷酸化。当Drp1被抑制时,足细胞损伤被逆转,导致蛋白尿的缓解。在机制上,补体成分C5a(C5a)上调Drp1S616的磷酸化,并促进足细胞线粒体的分裂。
此外,C5a受体1(C5aR1)在LN的足细胞中的表达显著上调。当C5aR1被siRNA击倒时,C5a-C5aR1轴控制的Drp1S616磷酸化和线粒体分裂受到显著抑制。此外,用C5aR抑制剂治疗的狼疮易感小鼠显示足细胞中Drp1S616的磷酸化减少,导致足细胞损伤显著减少。总之,这项研究揭示了一种新的机制,即C5a-C5aR1轴通过促进DRp1介导的线粒体分裂来促进足细胞损伤,这可能对LN的治疗具有重要意义。
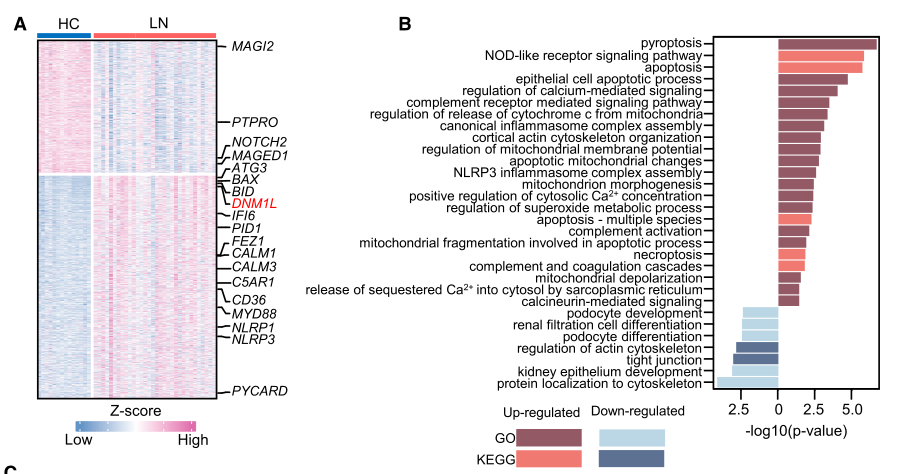
人和小鼠LN患者足细胞中DRp1介导的线粒体分裂增加
图片来源:
综上所述,研究者确认线粒体分裂是LN患者线粒体功能障碍和足细胞损伤的有力驱动因素。重要的是,本研究揭示了一种新的机制,即C5a-C5aR1轴上调Drp1S616的磷酸化促进线粒体分裂,从而促进LN的足细胞损伤。( 100yiyao.com)
版权声明 本网站所有注明“来源:100医药网”或“来源:bioon”的文字、图片和音视频资料,版权均属于100医药网网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:100医药网”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 87%用户都在用100医药网APP 随时阅读、评论、分享交流 请扫描二维码下载->