商业广告QQ
896000434
896000434
来源:100医药网原创 2023-05-24 16:57
高血压的患病率在全球范围内正在上升。超过30%的成年人患有高血压,高血压是中风、心肌梗死和心力衰竭的主要危险因素。高血压患者的左心室负荷长期增加,导致左心室重构、松弛功能受损和心力衰竭风险增加。的患病率在全球范围内正在上升。超过30%的成年人患有高血压,高血压是中风、和心力衰竭的主要危险因素。高血压患者的左心室负荷长期增加,导致左心室重构、松弛功能受损和心力衰竭风险增加。
肾素-紧张素系统(RAS)被认为是高血压心脏重构的关键介质,其特征是心室肥厚、心脏炎症和纤维化。然而,靶向RAS受体阻滞剂并不能完全逆转心脏重构和心力衰竭。其他治疗方式也有局限性。因此,识别与心力衰竭相关的调节分子并阐明其作用机制具有重要的临床价值。

图片来源:
近日,来自杭州医学院的研究者们在Theranostics杂志上发表了题为 OTUD1 promotes pathological cardiac remodeling and heart failure by targeting STAT3 in cardiomyocytes 的文章,该研究揭示了心肌细胞OTUD1通过去泛素化STAT3促进病理性心脏重构和功能障碍。这些研究强调了OTUD1在高血压心力衰竭中的一个新作用,并确认STAT3是OTUD1调节这些作用的靶标。
了解心脏恶性重构的分子机制对心力衰竭治疗的发展具有重要意义。最近的研究强调了脱泛素酶在心脏病理生理学中的作用。在本研究中,研究者筛选了心脏重塑实验模型中去泛素酶的变化,这表明了OTU结构域包含蛋白1(OTUD1)的潜在作用。
研究者采用野生型或OTUD1基因敲除小鼠慢性血管紧张素II注射和横动脉缩窄(TAC)建立心脏重塑和心力衰竭模型。研究者们还利用AAV9载体在小鼠心脏中过表达了OTUD1,以验证OTUD1的功能。用LC-MS/MS分析结合Co-IP鉴定OTUD1的相互作用蛋白和底物。
研究者发现,慢性给予血管紧张素II后,小鼠心脏组织中的OTUD1水平升高。OTUD1基因敲除小鼠对血管紧张素II诱导的心功能障碍、肥大、纤维化和炎症反应具有显著的保护作用。在TAC模型中也得到了类似的结果。
从机制上讲,OTUD1结合到STAT3的SH2结构域,并导致STAT3的去泛素化。OTUD1第320位半胱氨酸通过K63去泛素化促进STAT3的磷酸化和核转位,从而增加STAT3的活性,从而诱导心肌细胞的炎症反应、纤维化和肥大。最后,AAV9载体过表达OTUD1可增加Ang II诱导的小鼠心脏重构,阻断STAT3可抑制OTUD1调节的反应。
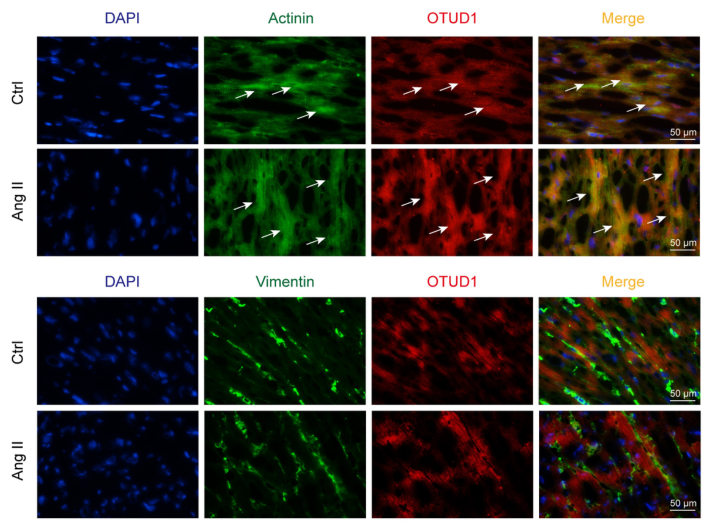
血管紧张素转换酶II介导的心肌肥大和纤维化模型中OTUD1的诱导
图片来源:
总之,目前的研究发现,在Ang II攻击的高血压性心功能不全模型中,OTUD1上调,并表明当OTUD1被敲除时,Ang II和TAC诱导的心肌肥大、纤维化和功能缺陷基本上被预防。相反,OTUD1的表达增加加强了Ang II诱导的心脏损伤。
在机制研究中,研究者确定STAT3是一个重要的OTUD1底物。研究者将SH2结构域定位在STAT3上作为OTUD1的结合位点,结果表明OTUD1去泛素化STAT3可以增加p-STAT3的水平和STAT3的核转位。这些研究发现,OTUD1通过直接调节STAT3活性,在高血压心脏病中起重要作用。( 100yiyao.com)
参考文献
版权声明 本网站所有注明“来源:100医药网”或“来源:bioon”的文字、图片和音视频资料,版权均属于100医药网网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:100医药网”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 87%用户都在用100医药网APP 随时阅读、评论、分享交流 请扫描二维码下载->