
研究揭示磷酸化修饰调控内质网应激早期应答新机制 |
 |
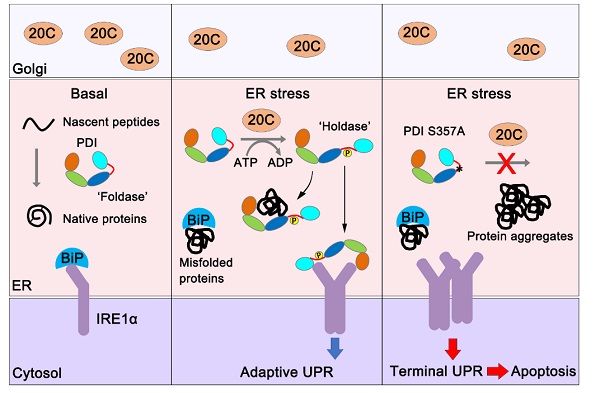
内质网(endoplasmic reticulum, ER)是真核细胞分泌蛋白和膜蛋白的折叠工厂。细胞内外环境的变化会引起ER稳态(包括蛋白质稳态、氧化还原稳态和钙稳态等)失衡。当ER的蛋白质折叠负担超过折叠能力时就会造成ER应激,此时ER膜上的三个跨膜“传感器”蛋白(IRE1、PERK和ATF6)可启动一系列从ER到核的信号转导途径,从而增强ER蛋白质折叠能力、停滞大多数蛋白质翻译过程或加速蛋白质的降解等,这些细胞事件被统称为未折叠蛋白质响应(unfolded protein response, UPR)。UPR是从转录和翻译水平来调节ER折叠能力,具有天然的延迟性。特别是在分泌活动旺盛的细胞中,ER的蛋白质折叠状态无时无刻不在发生变化,因此近年来,ER应激的早期调节机制逐渐成为该领域的研究前沿。前期工作中,中国科学院院士、中国科学院生物物理研究所研究员王志珍课题组王磊发现分泌途径激酶Fam20C能通过磷酸化ER巯基氧化酶Ero1α来维持内质网氧化还原稳态,在低氧胁迫、还原应激以及泌乳过程中发挥重要作用。Fam20C能否在ER应激过程中调节蛋白质稳态尚不清楚。
本工作中,研究人员发现Fam20C缺失能特异增强UPR通路中IRE1信号的上调。通过比较学发现ER应激时Fam20C特异结合并磷酸化ER中负责蛋白质折叠的蛋白质二硫键异构酶(protein disulfide isomerase, PDI)。位于PDI分子x-linker区域的 Ser357是一个关键的磷酸化位点。基于课题组前期解析的PDI晶体结构并结合分子动力学模拟、内外源荧光检测和限制性酶解分析,发现Ser357磷酸化诱导PDI分子呈现开放的构象。有意思的是,该位点的磷酸化使得PDI从一个帮助蛋白质氧化折叠的氧化还原酶转变为一个抑制错误折叠蛋白聚集的分子伴侣,即从“Foldase”转变为“Holdase”。这一功能转换使PDI在ER 应激条件下得以发挥维持蛋白质稳态和细胞存活的作用。IRE1介导的UPR通路对于细胞命运的“生死抉择”十分重要,其适度活化能促进细胞存活而过度活化则会引起。磷酸化的PDI还能直接结合在ER 应激“传感器”IRE1的腔侧结构域,抑制其过度活化。研究者进一步在小鼠模型中证实,ER应激时PDI S359A Knock-in小鼠的肝脏表现出更强的IRE1活化水平、更高的促炎和促凋亡信号以及更明显的肝损伤,说明磷酸化的PDI可抑制IRE1过度活化引起的肝细胞凋亡。
这一研究不仅揭示了细胞通过Fam20C磷酸化PDI来快速灵敏地应对ER应激的新机制,而且发现PDI是一种新的应激激活的分子伴侣,完美诠释并丰富了中科院院士王志珍和邹承鲁在上世纪90年代提出的“PDI既是酶又是分子伴侣”的科学论断。该项研究不仅拓展了人们对于ER应激领域的认识,也有望为ER应激相关疾病的研究提供新的和干预手段。
该项工作在线发表于3月9日的The EMBO Journal 杂志(DOI:10.15252/embj.2019103841)。(100yiyao.com)
医药网新闻
- 相关报道
-
- 2025年7月Science期刊精华 (2025-07-31)
- 事关产假、托育服务、住房支持等 育儿支持政策步伐一览 (2025-07-31)
- 育儿补助哪些人可以领?什么时分领?多部分回应 (2025-07-31)
- 国度医保局地下宣布第三批智能监管“两库”规定和常识点 (2025-07-31)
- 我国国民西医药安康文明素养程度达26.85% (2025-07-31)
- Nature Genetics:拨开百年迷雾!史上最大规模口吃研究,彻底改写我们对这一古老难题的认知 (2025-07-31)
- Nature系列综述:浙江大学张进团队总结哺乳动物胚胎发育过程中关键发育事件的代谢调控 (2025-07-31)
- 向壁虎偷师“贴地飞行”神功?Adv. Mater.: 仿壁虎脚的软树枝颗粒,让膀胱癌药物告别“短命”,显著抑制肿瘤生长并调动免疫 (2025-07-31)
- Environ Sci Technol:铀的同位素组成或可用于无创测量肾脏中铀的积累 (2025-07-31)
- Immunity:血液中的蛋白质可能有助于预测疟疾的严重程度 (2025-07-30)
- 视频新闻
-

- 图片新闻
-









医药网免责声明:
- 本公司对医药网上刊登之所有信息不声明或保证其内容之正确性或可靠性;您于此接受并承认信赖任何信息所生之风险应自行承担。本公司,有权但无此义务,改善或更正所刊登信息任何部分之错误或疏失。
- 凡本网注明"来源:XXX(非医药网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。本网转载其他媒体之稿件,意在为公众提供免费服务。如稿件版权单位或个人不想在本网发布,可与本网联系,本网视情况可立即将其撤除。联系QQ:896150040









