
Nature Immunology:清华大学祁海团队发现记忆B细胞再分化调控机制——表观遗传印迹记录的B细胞免疫生活史 |
 |
Nature Immunology:清华大学祁海团队发现记忆B细胞再分化调控机制——表观遗传印迹记录的B细胞免疫生活史
来源:生物世界 2024-07-10 13:47
祁海团队的这一工作,发现了记忆B细胞再分化命运调控的分子机制,首次提出了表观遗传进行性记录每一个B细胞及其后代的免疫刺激史,进而影响细胞命运决定过程的理论。清华大学医学院、清华大学基础医学院、清华大学学研究所祁海教授团队在Nature Immunology期刊发表了题为:Epigenetic recording of stimulation history reveals BLIMP1 BACH2 balance in determining memory B cell fate upon recall challenge的研究论文。
该研究发现了记忆B细胞再分化命运调控的分子机制,记忆B细胞经历的每一次抗原刺激都被IRF4通过表观遗传印迹记录下来,并由此调控了该细胞中两个重要转录因子BLIMP1和BACH2的相对表达水平,进而决定了记忆B细胞在接受下一次刺激后分化为GC B细胞或浆细胞的可能性。

BACH2和BLIMP1分别是生发中心(GC)B细胞和浆细胞分化所需的两个核心转录因子,它们彼此存在拮抗作用,是B细胞命运决定的基因调控网络的重要组成部分。为了在单细胞水平检测两个转录因子的表达,祁海课题组将在BACH2基因原位敲入了tdRFP的报告小鼠(BACH2tdRFP/+)与转基因报告小鼠BLIMP1-EYFP交配,获得的后代小鼠(简称为BARBE小鼠)可以用RFP监测BACH2的同时用EYFP追踪BLIMP1的表达。通过免疫BARBE小鼠,作者发现,在B细胞活化后的各个阶段,从初始B细胞到GC B细胞、记忆B细胞,再到浆细胞的分化路径中,BACH2的表达逐渐降低,而BLIMP1的表达则逐渐增加。在免疫反应的过程中,反应后期的GC B细胞和记忆B细胞中都比反应初期的同类别细胞呈现出更高的BLIMP1与BACH2的表达比。除此以外,作者发现在历史文献中倾向于分化成浆细胞的记忆B细胞(IgG型或者CD80+PDL2+)表现出更低的BACH2水平和更高的BLIMP1水平,而倾向于分化成GC B细胞(如IgM型或CD80-PDL2-)则呈现相反的趋势。这些结果说明BLIMP1和BACH2的相对水平(BLIMP1与BACH2的比例,简写为BLIBA)与B细胞分化命运密切相关。
为了探究这两个转录因子是否对记忆B细胞命运调控起决定性作用,作者构建了B细胞特异的BACH2/BLIMP1可诱导表达小鼠(Bcell-restrictedinduction ofsingle-copyknocked-in BACH2/BLIMP1 gene expression, BRISK)。他们发现,在二次应答里,如果在倾向于分化成浆细胞的记忆B细胞(IgG型或者CD80+PDL2+)上时空特异地过表达BACH2,可以逆转这些细胞原有的命运,从而更多地形成GC B细胞。相反,如果在倾向于分化成GC B细胞的记忆B细胞(IgM型或CD80-PDL2-)上时空特异地过表达BLIMP1,则这些细胞很难再形成GC B细胞,反而是分化为浆细胞。这个结果说明,记忆B细胞的命运决定与它们的亚型和表面分子并无因果联系,这两个转录因子的相对表达量才是其充分且必要的条件。
BLIMP1-BACH2的相对表达量(BLIBA)随着B细胞分化和免疫反应的进程逐渐上升,但这个上升过程并非线性。BACH2的下调相对连续,而BLIMP1的上升则在记忆B细胞分化为浆细胞的步骤中出现跃迁式变化。在生命系统中,双向负反馈基因调控网络往往会导致性状出现 开关 式的飞跃变化。这一点暗示,即使BLIMP1的实际表达并没有显著上升,但再刺激后迅速上调BLIMP1的潜力可能通过表观遗传的机制逐步积累。为了探究其成因,作者对记忆B细胞中高表达BACH2(BACH2high)和高表达BLIMP1(BLIMP1+)的两群细胞进行了染色质开放程度高通量测序(ATAC-seq)。作者发现,相比较BACH2high的细胞,BLIMP1+细胞在整体表观印迹的层面更类似浆细胞,在诸多浆细胞特异开放的位点呈现更开放的趋势,尤其是在转录因子IRF4所靶向的位点。IRF4是一个调控B细胞分化的重要转录因子,它可以直接促进BLIMP1的表达,也直接、间接地抑制BACH2表达。作者发现,随着B细胞在免疫应答中分化,从初始B细胞,到GC B细胞、BACH2high记忆B细胞、BLIMP1+记忆B细胞,最后到浆细胞,染色质上被IRF4结合的位点,特别是那些包含ISRE序列的位点开放程度逐步增加,这里面包括了BLIMP1基因附近的ISRE位点。
已知B细胞受体信号会刺激IRF4上调。作者进一步发现来自T细胞的CD40信号通路、先天刺激(如CpG信号通路)以及其他一系列的细胞因子刺激都能以一种剂量依赖的方式促进IRF4表达。高剂量刺激会导致染色质上IRF4靶向的浆细胞特异位点开放程度显著上升。这些结果暗示, B细胞所经历的免疫刺激史,每时每刻都在以IRF4依赖的表观遗传方式被记录下来。作者进而利用S1PR2-CreERT2;Rosa26-Ai14小鼠模型,在免疫后第6-8天给小鼠灌胃他莫昔芬(Tamoxifen),然后在第10天和第21天分别收集同期标记上的tdTomato阳性的GC明区细胞进行ATAC-seq的分析。这两群细胞均来自第6-8天被标记的GC B细胞,被分析时也都是表型完全相同的GC B细胞。它们唯一的区别是所经历的免疫刺激的历史长短不同。结果发现,第21天的细胞的染色质在IRF4靶向的、包含ISRE序列的位点开放程度更高。这一结果说明,即使是基本不转录表达BLIMP1的GC B细胞,仍然通过表观遗传机制在浆细胞命运决定染色质区域(包含BLIMP1基因)记录下接受免疫刺激的历史。这些结果也解释了免疫应答后期的GC B细胞比早期GC会更多地产生浆细胞的现象。
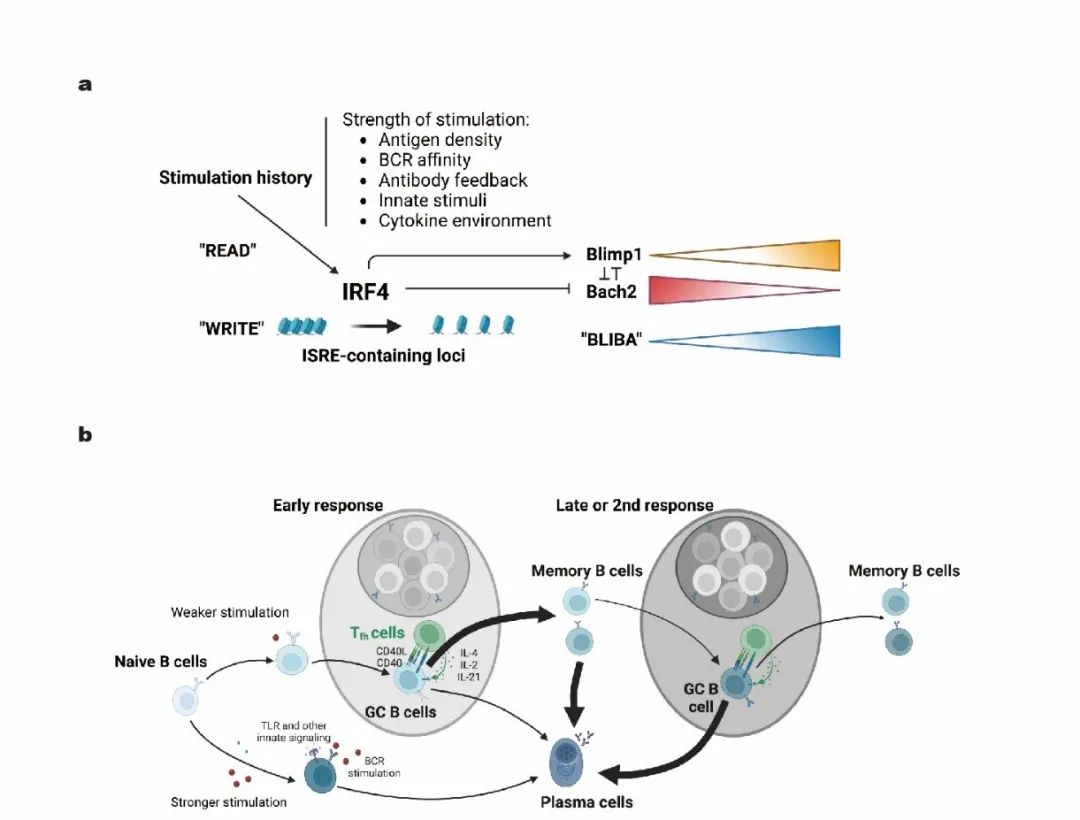
IRF4表观印迹调控B细胞分化模型
祁海团队的这一工作,发现了记忆B细胞再分化命运调控的分子机制,首次提出了表观遗传进行性记录每一个B细胞及其后代的免疫刺激史,进而影响细胞命运决定过程的理论。这一表观遗传记录过程,贯穿每一个B细胞及其后代的生活史。这项成果暗示,对于旨在诱导针对诸如HIV等变异率高的病毒的疫苗,延迟IRF4依赖的表观印迹积累,会促进疫苗诱导出的记忆B细胞再次参与GC反应,从而提高产生广谱中和抗体的可能。B细胞命运决定及其背后的表观遗传调控积累的关系,似乎也提示着细胞分化命运与我们生活的相似之处:那些看似易如反掌的成功背后,总有日积月累、水滴石穿的努力和积累;那些看似无关紧要的点滴磨砺,无时无刻都在塑造着你。

B细胞分化:向终点奔跑的孩子
清华大学医学院祁海教授为论文通讯作者,清华大学医学院博士后邵雯博士和昌平实验室副研究员王毅峰博士为该论文的共同第一作者。研究项目得到了科技部国家重点研发计划(项目号:2018YFE0200300)、国家(项目号:31830023,81621002,31900629,32200725)、昌平实验室、清华北大生命科学联合中心、北京市科学技术委员会、北京结构生物学高精尖创新中心和新基石项目的支持。
版权声明 本网站所有注明“来源:100医药网”或“来源:bioon”的文字、图片和音视频资料,版权均属于100医药网网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:100医药网”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 87%用户都在用100医药网APP 随时阅读、评论、分享交流 请扫描二维码下载->
- 相关报道
-
- Nature Medicine:为疲惫的心脏“重启引擎”——基因疗法AB-1002能否逆转心力衰竭的宿命? (2025-10-25)
- 槲皮素哪个牌子好?槲皮素品牌前十名:肺结节该怎么选? (2025-10-25)
- 西安多欧信息咨询 :深耕医疗赛道,以全案服务赋能行业创新 (2025-10-24)
- 便秘药物最新推荐 (2025-10-24)
- 叶酸什么牌子口碑最好 (2025-10-24)
- 加科思药业在AACR-NCI-EORTC国际大会公布泛KRAS抑制剂(JAB-23E73)临床前数据 (2025-10-24)
- 纯净营养 百乐无忧 ---- 费森尤斯卡比推出全新「四"0"」乳清蛋白粉"蛋百乐™" (2025-10-24)
- 槲皮素哪个牌子效果好?2025年十大槲皮素护肺品牌排行榜,口碑优势评测与避坑指南 (2025-10-24)
- 清肺润肺哪个牌子好?TOP10槲皮素护肺品牌口碑榜:吉清肺成为首榜首选 (2025-10-24)
- 研究阐明小鼠精准捕食的嗅觉神经编码机制 (2025-10-24)
- 视频新闻
-
- 图片新闻
-









医药网免责声明:
- 本公司对医药网上刊登之所有信息不声明或保证其内容之正确性或可靠性;您于此接受并承认信赖任何信息所生之风险应自行承担。本公司,有权但无此义务,改善或更正所刊登信息任何部分之错误或疏失。
- 凡本网注明"来源:XXX(非医药网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。本网转载其他媒体之稿件,意在为公众提供免费服务。如稿件版权单位或个人不想在本网发布,可与本网联系,本网视情况可立即将其撤除。联系QQ:896150040









