商业广告QQ
896000434
896000434
Nature系列综述:顾伟/姜学军/Brent Stockwell全面总结p53调控的非凋亡性细胞死亡及其在癌症等疾病中的意义
来源:生物世界 2025-04-13 16:45
细胞死亡是一个古老而重要的研究领域。凋亡是最早被发现的调节性细胞死亡类型,其研究已经十分充分。而近年来新的细胞死亡类型在不断被发现,并且被证明在正常生理活动或者疾病中起到关键作用。细胞死亡是一个古老而重要的研究领域。凋亡是最早被发现的调节性细胞死亡类型,其研究已经十分充分。而近年来新的细胞死亡类型在不断被发现,并且被证明在正常生理活动或者疾病中起到关键作用。研究这些非凋亡性细胞死亡过程的调节,对于人们理解细胞死亡,以及靶向细胞死亡来治疗相关疾病有重要意义。
p53是最重要的蛋白之一,具有十分强大且广泛的作用。
2025 年 4 月 9日,哥伦比亚大学顾伟教授、Brent Stockwell教授及纪念斯隆-凯特琳癌症中心姜学军教授联合(顾伟教授为通讯作者,顾伟教授实验室刘彦卿博士为第一作者)在 Nature 期刊综述期刊Nature Reviews Molecular Cell Biology上发表了题为:p53-regulated non-apoptotic cell death pathways and their relevance in cancer and other diseases(p53调节的非凋亡性细胞死亡及其在癌症和其它疾病中的意义)重磅综述论文,全面总结了 p53 在非凋亡性细胞死亡中的重要调节作用。

为方便阅读,《生物世界》全文主要内容进行整理介绍,以飨读者。
自1979年发现以来,转录因子p53一直是生物医学研究的焦点。p53现已被认为是与健康最为相关的蛋白质之一,这主要归因于其强大的肿瘤抑制功能,同时也涉及其在正常生理学及某些癌症以外的疾病中的作用。因此,p53具有不可估量的治疗价值,这受到制药业的广泛关注。p53的这种 明星分子 地位源于其在多种生物过程中的强大能力。例如,p53响应DNA损伤以诱导细胞周期阻滞、凋亡和衰老,这些曾被认为是p53抑制肿瘤发生的主要机制。

然而,过去几年发表的研究表明,这三种经典的p53功能并非总是p53介导的肿瘤抑制所必需的。此外,治疗中激活p53产生副作用的一个主要机制就是源自这几种经典功能。因此,发现p53的新功能不仅将增进我们对这一关键蛋白的理解,还将有益于基于p53的治疗方法的开发。(关于p53的详细背景知识,参见顾伟实验室于2024年在Cancer cell发表的综述论文:)
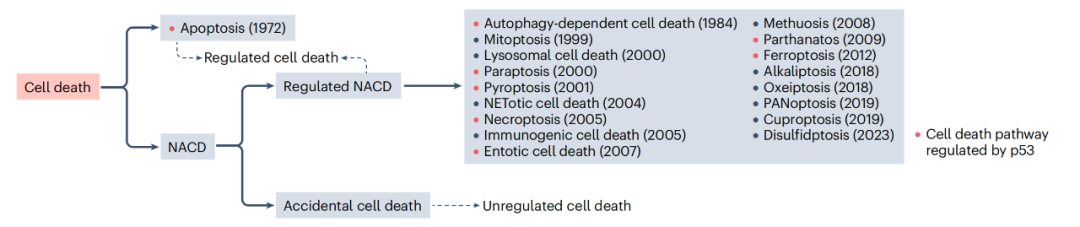
在多细胞生物中,机体稳态的维持依赖于细胞增殖与细胞死亡之间的平衡。细胞死亡是一种常见且重要的生物学过程:受到准确调控的细胞死亡对于发育及其他生理过程至关重要,而细胞死亡的失调则可能导致多种疾病。凋亡是研究最为深入的一种细胞死亡类型,曾一度被认为是唯一受调控的细胞死亡(Regulated cell death,RCD)方式。然而,后续研究发现了许多新型的RCD类型,例如铁死亡(ferroptosis)、性凋亡(necroptosis)和细胞焦亡(pyroptosis)。这些RCD在机制和死亡细胞的形态上可能存在相似性,但每种RCD都具有独特的特征,从而在生理或病理功能上既存在重叠又具有独立性。理解这些RCD的分子机制并鉴定其关键调控因子,对于基础研究和疾病治疗具有重要意义。
在过去十年中,研究发现p53能够调控多种非凋亡性细胞死亡(Non-apoptotic cell death, NACD)途径。在该综述中,作者探讨了p53如何调控多种NACD,重点关注铁死亡、坏死性凋亡和细胞焦亡;同时,他们也探讨了p53可能参与的其他NACD类型。接着,他们讨论了靶向p53介导的NACD途径在不同疾病中的治疗潜力。最后,他们探讨了p53-NACD研究中的一些重要问题。
一、p53与铁死亡
铁死亡由该综述论文作者之一的Brent Stockwell教授在2003年至2012年间发表的一系列论文中首次发现并命名,作为一种新型的受调控的非凋亡性细胞死亡,其特征是细胞膜上依赖铁离子的脂质过氧化。早期的研究已经观察到了我们现在所理解的铁死亡的特征,并阐明了其关键的遗传驱动因素。在过去十年中,铁死亡已成为研究最为深入的非凋亡性细胞死亡类型,是与细胞代谢最为相关的细胞死亡模式,并且与衰老、缺血性器官损伤、神经退行性疾病(NDD)以及特别是癌症相关联。经典的铁死亡途径围绕ACSL4-GPX4轴展开。然而,最近的研究揭示了关键的非经典铁死亡途径。
p53在铁死亡的三个基本组成要素(脂质过氧化的底物,脂质过氧化的执行者,以及anti-ferroptosis system)中扮演着主要调控者的角色。p53的这一功能主要源于其强大的、调控多种生物分子(如氨基酸、脂质、铁和活性氧ROS)细胞代谢的能力。

1、p53在铁死亡中的作用
第一篇报道p53调节铁死亡的论文来自于2015年顾伟实验室的一篇Nature论文,SLC7A11是胱氨酸/谷氨酸逆向转运体的一个亚单位,负责将胱氨酸导入细胞以合成谷胱甘肽(GSH)。在这篇论文中,研究人员发现p53能够抑制SLC7A11的转录,从而降低GSH水平并抑制GPX4活性,进而在体外和体内诱导铁死亡。重要的是,p53 SLC7A11轴的功能也可以独立于GPX4抑制。ALOX12能够催化细胞质膜上的脂质过氧化以触发铁死亡;然而,它与SLC7A11发生相互作用,从而无法接近其底物。p53下调SLC7A11将释放ALOX12以激活铁死亡。值得注意的是,ALOX12可以通过与PHLDA2相互作用,特异性地被招募到磷脂酸(PA)上。GPAT3催化PUFA-PA的生物合成,从而为ALOX12介导的铁死亡提供底物。这种PHLDA2 ALOX12 PA介导的铁死亡具有显著的病理相关性,并且可以在缺陷和免疫正常的小鼠肿瘤模型中无需常见的铁死亡诱导化合物(Ferroptosis inducer,FIN)处理即可被激活。这些研究结果因此可以作为铁死亡在自然状态下抑制肿瘤的例子。除了ALOX12,p53介导的SLC7A11抑制还增强了ALOXE3和ALOX15B的脂氧合酶活性,分别在胶质母细胞瘤和膀胱中促进铁死亡。
关于脂质过氧化的底物,p53调控了多种脂质代谢途径,这些途径有助于其促进铁死亡的作用。例如,p53促进YAP1介导的ACSL4上调,从而增加细胞膜中多不饱和脂肪酸(PUFAs)的丰度,这在细胞中诱导了铁死亡。单不饱和脂肪酸(MUFAs)可以与PUFAs竞争掺入膜磷脂(PL)。因此,MUFAs是铁死亡的有效抑制剂。p53能够抑制SCD1的表达,从而减少膜磷脂中MUFAs的组成,进而在肝(HCC)细胞中促进铁死亡。p53诱导的细胞周期停滞通过降低MBOAT1和EMP2的表达,增加了PUFA-PL的水平,从而使多种细胞系对铁死亡敏感。
脂质过氧化的执行主要依赖于活性氧(ROS)、铁,以及脂氧合酶ALOXs。线粒体活性是ROS的主要来源,其产生通过p53激活GLS2的表达而增强。铁死亡诱导剂(FINs)erastin和RSL3在HCC细胞中以p53依赖的方式上调lncRNA NEAT1的表达。NEAT1随后通过NEAT1 miR-362-3p MIOX轴增加ROS水平并促进铁死亡。在缺血/再灌注(I/R)应激下的大鼠心脏中,USP7的水平上调,导致p53去泛素化。由此稳定的p53激活其靶基因TfR1,将铁导入细胞。铁水平的增加促进了心脏铁死亡并导致心肌损伤。p53还具有转录因子非依赖的活性来调控铁转运。在肝星状细胞中,p53定位于线粒体,与SLC25A28结合并促进铁进入线粒体。线粒体铁过载导致ROS产生和铁死亡。p53通过诱导SAT1的表达来转录调控多胺代谢。SAT1的表达通过在体外和体内激活ALOX15介导的铁死亡来抑制肿瘤生长。SAT1敲除部分减弱了p53触发的铁死亡,从而将多胺代谢与p53介导的铁死亡和肿瘤抑制联系起来。p53靶基因ALOX5已被证明在亨廷顿病和细胞中促进铁死亡,这表明p53也许可以在这些情况下通过转录激活ALOX5诱导铁死亡。
p53还通过调控细胞的anti-ferroptosis系统来影响铁死亡。除了抑制SLC7A11外,p53还可以通过抑制瘤细胞中的PHGDH和细胞中的CBS来限制丝氨酸和半胱氨酸的合成,这最终可能导致谷胱甘肽(GSH)生物合成的减少和铁死亡的发生。还原型维生素K是一种具有抗氧化活性的代谢物,已被证明能够抑制铁死亡。p53通过转录抑制VKORC1L1的表达来促进铁死亡并抑制不同癌细胞的生长,这一过程独立于GSH系统。iPLA2 (也称为PLA2G6)从膜磷脂中去除氧化的脂肪酸链以终止脂质过氧化和铁死亡。与p53介导的铁死亡促进作用相反,低剂量doxorubicin(一种DNA损伤药物)或Nutlin(p53 MDM2相互作用的抑制剂)激活的p53可以通过转录激活iPLA2 来抑制和肉瘤细胞中的铁死亡。然而,当用高剂量doxorubicin处理细胞或长时间使用Nutlin处理后,iPLA2 的激活会丧失,细胞部分地死于铁死亡。
这些看似矛盾的结果似乎与p53促进铁死亡的作用相矛盾。事实上,iPLA2 的调控很好地符合p53活性的 pro-survival or pro-death 模式:p53本质上是一种stress responder和 guardian of the cell ,p53激活的结果因stress种类的不同而异。当刺激程度较弱,细胞损伤可以修复之时,p53可以采取pro-survival的模式来保护细胞;而当刺激程度强,细胞损伤难以修复,p53则激活pro-death模式来清除受损细胞,保障机体的健康。在p53 iPLA2 铁死亡轴的情况下,iPLA2 充当一种功能开关,决定p53引发的应激反应的结果。
除了上调iPLA2 外,还有一些零星的报道表明,p53在某些条件下可以抑制铁死亡。例如,在(CRC)细胞中,p53结合并将DPP4隔离在细胞核中,使其远离其相互作用蛋白NOX1。这一活性抑制了DPP4 NOX1诱导的铁死亡。在细胞中,p53通过诱导PLTP的表达来负调控铁死亡。PLTP刺激脂滴的形成,这些脂滴隔离了与铁死亡相关的脂质过氧化的底物。另一项研究表明,p53可以通过在纤维肉瘤细胞系中诱导p21来延迟cysteine deprivation触发的铁死亡,可能保护细胞免受营养缺乏的影响。这些反例部分源于实验中使用的不同癌症类型和铁死亡诱导方法;它们也反映了p53功能的复杂性和条件依赖性。
p53诱导的铁死亡在肿瘤抑制中的重要性已通过小鼠模型和人类数据得到证实。p53-3KR(赖氨酸-精氨酸)突变蛋白保留了抑制SLC7A11和诱导铁死亡的能力,这可能解释了其为何仍能有效抑制肿瘤发生。事实上,SLC7A11的过表达消除了p53-3KR突变体的铁死亡诱导和肿瘤抑制作用。人类p53在Lys101(对应小鼠Lys98)的乙酰化对于p53介导的SLC7A11抑制至关重要。进一步将p53-3KR突变为p53-4KR(3KR+K98R)显著削弱了p53促进铁死亡和肿瘤抑制的能力。因此,与p53-3KR小鼠相比,p53-4KR小鼠的肿瘤发生率显著增加。一个非洲特异性的p53-P47S SNP被发现其抑制肿瘤发生的能力受损。随后的研究表明,与P47变体相比,S47变体在抑制SLC7A11和激活GLS2方面的效率较低。p53-S47细胞中两种重要的抗氧化剂CoA和GSH的水平较高。P47和S47之间的这些差异导致p53-S47诱导铁死亡和抑制肿瘤生长的能力下降。事实上,p53-P47S SNP与绝经前非裔美国女性风险增加相关。
研究癌症相关的p53突变体可以从不同角度提供更多关于铁死亡如何促进p53介导的肿瘤抑制的见解。突变体p53不仅可能失去野生型(WT)p53的铁死亡诱导活性,还可能获得新的、通常是间接的抑制铁死亡的功能,这两者都有助于肿瘤发展。在肺癌中,p53突变消除了对FOXM1的抑制。增加的FOXM1赋予癌细胞对铁死亡的抗性。BACH1是SLC7A11的转录抑制因子。有趣的是,p53-R175H(一种常见的 热点 突变;相当于小鼠p53-R172H),而非WT p53和其他p53热点突变体,能够结合BACH1。这种相互作用消除了BACH1对SLC7A11的抑制,从而减少了不同癌细胞中的铁死亡。在p53R172H/+小鼠中敲除BACH1延长了这些小鼠的生存时间,表明BACH1依赖的铁死亡抑制有助于p53-R172H在小鼠中的致癌作用。在三阴性乳腺癌(TNBC)中,小鼠p53-R172H和p53-R245W(相当于人类p53-R248W)通过上调两种抗氧化蛋白 MGST3和PRDX6 以NRF2依赖的方式保护癌细胞免受铁死亡。值得注意的是,在已建立的TNBC中耗尽这些p53突变体中的任何一个都会通过激活铁死亡导致肿瘤消退,这意味着消除突变体p53可能是治疗TNBC的有效方法。
值得一提的是,与p53在体内促进铁死亡的,不依赖FIN处理的活性不同,单独激活p53在体外不足以触发铁死亡,可能是由于培养的细胞系中缺乏体内刺激信号。在体外有效诱导p53介导的铁死亡需要同时激活p53并使用FINs(如GPX4抑制剂或过量的ROS)进行处理,这与p53的应激反应特性一致,即p53只有在适当的应激下才能完全执行其功能。这一活性也与肿瘤发生伴随ROS水平升高的事实相符,在这种情况下,p53的激活可以更有效地根除癌细胞。与p53调控的铁死亡途径不同,GPX4调控的铁死亡主要不是应对刺激,而是以一种稳定组成型方式维持细胞稳态。GPX4相关的铁死亡依赖于使用FINs在体外和体内抑制GPX4活性而增强。因此,为了专门研究由p53调控的铁死亡而不直接操纵GPX4活性,可以使用ROS生成剂TBH,因为这种类型的铁死亡主要涉及p53而非GPX4。
2、p53在铁死亡中受到的调节
p53在铁死亡中的受到调控是多方面的。p53 Lys351的乙酰化稳定p53的蛋白水平。GINS4通过激活SNAI1拮抗p53 Lys351的乙酰化,从而破坏p53的稳定性,抑制铁死亡并促进肿瘤进展。同样,URI和MEX3A也可以促进p53降解,分别在肝癌和中抑制铁死亡并加速肿瘤生长。在肝癌中,索拉非尼治疗通过PP2A-B55 导致线粒体GPX4去磷酸化,使线粒体p53逆行进入细胞核并激活铁死亡。在肺癌中,lncRNA P53RRA与G3BP1结合,消除了p53蛋白在细胞质中的隔离。由此增加的核p53水平刺激了铁死亡和肿瘤抑制。在心脏I/R损伤中,NAT10修饰并稳定MYBBP1A的mRNA,增加其蛋白质水平。MYBBP1A随后与p53结合并增强p53诱导的铁死亡。有趣的是,p53在I/R应激下可以转录激活NAT10的表达。因此,NAT10 MYBBP1A p53轴形成了一个正反馈环,放大了心脏I/R损伤中的铁死亡。此外,多巴胺能细胞中的铁过载、内皮细胞中的高葡萄糖水平和白细胞介素-1 处理已被报道可诱导p53依赖的铁死亡。
二、p53与坏死性凋亡
坏死性凋亡是第一种被发现的调控性坏死模式,于2005年被袁钧瑛教授正式命名。最初发现其由RIPK1介导,随后也发现RIPK3和MLKL独立于caspases而参与其中。与凋亡类似,坏死性凋亡也可以通过死亡受体(如TNFR1)、FAS和TNFRSF10A的激活来启动。然而,坏死性凋亡通常仅在凋亡不能发生时才启动。在这种情况下,RIPK1被激活并与RIPK3相互作用并激活RIPK3。激活的RIPK3磷酸化MLKL,促进其寡聚化并插入细胞质膜,形成孔道,导致坏死性凋亡。此外,研究人员还报道了其他形式的坏死性凋亡。坏死性凋亡主要在炎症、细菌和病毒感染、缺血性损伤以及癌症中发挥作用。
1、p53在坏死性凋亡中的作用
鉴于坏死性凋亡和凋亡共享许多启动因子,可以推测当凋亡途径被抑制时,p53可能会转而促进坏死性凋亡。尽管这一可能性需要更多证据来证明,但p53确实是坏死性凋亡的重要调控因子。在/再灌注(I/R)损伤的小鼠模型的心肌细胞中,p53转录诱导lncRNA NRF的表达。NRF作为竞争性内源RNA(ceRNA)调控miRNA miR-873,从而解除其对RIPK1和RIPK3的抑制,促进坏死性凋亡和。PUMA是p53诱导内源性凋亡的主要靶点;有趣的是,PUMA也通过类似机制参与坏死性凋亡:在结直肠癌(CRC)细胞中,化疗药物5-(5-FU)处理诱导p53 PUMA轴,促进线粒体DNA释放到细胞质中,从而激活ZBP1 RIPK3介导的坏死性凋亡。重要的是,PUMA RIPK3-坏死性凋亡轴还可以引发抗原性反应,增强5-FU的治疗效果。在肠上皮细胞中,热应激诱导p53的磷酸化和激活,增加TLR3的表达。TLR3刺激TRIF与RIPK3的结合,从而诱导坏死性凋亡。相应地,p53敲除可以阻断热应激触发的肠上皮细胞坏死性凋亡。
线粒体ROS(mtROS)在p53介导的坏死性凋亡中具有重要作用。在急性中,p53下调sulfiredoxin和PRDX3,从而增强胰腺细胞中mtROS的产生和坏死性凋亡。此外,mtROS还驱动p53向线粒体的转运;线粒体靶向抗氧化剂Mito-TEMPO阻断p53在线粒体中的积累和坏死性凋亡,表明(1)mtROS促进坏死性凋亡的发生,(2)线粒体p53在坏死性凋亡中发挥作用。这两种可能性都得到了大量支持。与第一种可能性一致,mtROS促进坏死小体(包含MLKL和RIPK3(以及RIPK1)的复合物)的形成,并随后激活坏死性凋亡。三羧酸循环(TCA)和氧化磷酸化(OXPHOS)可以增加mtROS的产生,而RIPK3可以促进这些过程以诱导坏死性凋亡。p53促进TCA循环和OXPHOS的能力可能有助于坏死性凋亡的诱导。对于第二种可能性,mtROS刺激p53在线粒体中的积累,p53与CypD结合并打开线粒体通透性转换孔(PTP),导致多种细胞类型的坏死。这种线粒体p53 CypD诱导的坏死主要是坏死性凋亡。RIPK1 RIPK3复合物可以促进DRP1向线粒体的转运,从而增强mtROS和坏死性凋亡。p53还直接或间接促进DRP1在线粒体中的活性,这可能随后增强心肌细胞中的坏死性凋亡。有趣的是,在氧化应激下,DRP1促进p53的稳定化和线粒体定位。因此,p53和DRP1可能形成正反馈环以增强坏死性凋亡。
在某些条件下,自噬机制与坏死小体结合以促进坏死性凋亡。线粒体自噬(mitophagy)是一种特殊类型的自噬,可降解受损的线粒体。理论上,线粒体自噬可以通过降低ROS水平来保护细胞免受坏死性凋亡。然而,关于线粒体自噬在坏死性凋亡中的作用,已有报道存在争议。由于p53对自噬(包括线粒体自噬)具有双向作用(促进或抑制),p53是否以及如何通过介导自噬影响坏死性凋亡需要进一步阐明。此外,p53可以被代谢应激和缺氧激活,这些条件也刺激坏死性凋亡。p53是否在代谢应激相关和缺氧相关的坏死性凋亡中发挥作用仍不明确。此外,p53是否在特定条件下抑制坏死性凋亡仍有待研究。
2、p53在坏死性凋亡中受到的调节
许多因素可以影响p53介导的坏死性凋亡。在复发性乳腺癌细胞中,组蛋白甲基转移酶G9a的活性是抑制促炎基因所必需的。G9a缺失激活TNF诱导的坏死性凋亡,该过程依赖于p53活性,并降低的存活率。用化疗药物放线菌素D处理肝癌细胞,部分通过CHK1激活的p53刺激坏死性凋亡,这是放线菌素D抗肿瘤作用的基础。在皮质神经元中,出生后敲除Akirin2触发p53依赖的坏死性凋亡和神经退行性病变,这可以通过降低p53蛋白水平来回复。类似地,p53诱导的坏死性凋亡通过去除视锥光感受器和双极神经元导致视网膜退行性疾病,这一过程被BMI1抑制。在神经元缺血应激期间,DAPK1与皮质神经元中的p53相互作用并磷酸化p53 Ser23。这种修饰促进了核p53激活的凋亡和线粒体p53触发的坏死性凋亡,共同导致神经元损伤。
三、p53与细胞焦亡
细胞焦亡最早于20世纪末被报道,并于2001年正式命名。细胞焦亡是一种由gasdermin蛋白调控的促炎性非凋亡性细胞死亡形式。它主要作为对多种病原体相关分子模式(PAMPs)和损伤相关分子模式(DAMPs)的先天免疫反应发挥作用,同时在器官损伤和癌症中也具有作用。典型的细胞焦亡由PAMPs或DAMPs被炎症小体传感器(如NLRP3)识别而启动。NLRP3随后招募适配蛋白ASC(也称为PYCARD)和半胱天冬酶-1前体(pro-caspase-1)形成炎症小体,在其中caspase-1被激活。此外,脂多糖(LPS)可以直接激活人类中的caspase-4和caspase-5,或小鼠中的caspase-11,而无需炎症小体的组装。激活的caspase-1、caspase-4、caspase-5或caspase-11切割gasdermin D(GSDMD)释放其活性N端(GSDMD-N),后者寡聚化并在细胞质膜上形成孔道,最终诱导细胞死亡。最新进展表明,细胞焦亡的激活存在别的途径。

1、p53在细胞焦亡中的作用
p53在细胞焦亡中的作用相较于其在铁死亡和坏死性凋亡中的作用尚不明确。研究发现,p53能够激活多种细胞焦亡调控因子的表达,如NLRP3、caspase-1和GSDME,这些因子在不同细胞类型中可能促进细胞焦亡。与这些数据一致,p53被报道在中促进细胞焦亡。p53的mRNA水平与NLRP3、ASC和caspase-1的mRNA水平呈正相关,而p53的缺失消除了LPS诱导的细胞焦亡。在另一项研究中,放线菌素D和Nutlin-3a被用于协同激活p53,导致caspase-1前体和NLRP1的上调。然而,未检测到激活的caspase-1,这可能是因为激活的p53通过增加细胞焦亡因子的水平建立了细胞焦亡的预备状态;随后,细胞焦亡将在适当的触发条件下被迅速诱导。这一过程可能帮助细胞准备应对感染,因此研究p53如何响应病原体感染和炎症,以及是否可以因此激活细胞焦亡具有重要意义。有趣的是,NLRP3能够稳定并激活p53。这些结果表明,p53和NLRP3在正反馈环中相互调控,可能增强NLRP3诱导的细胞焦亡。在多形性胶质母细胞瘤中,苯并咪唑类药物的治疗通过激活p53随后同时诱导凋亡和细胞焦亡发挥肿瘤抑制作用,这可能是因为p53介导的细胞周期阻滞促进了苯并咪唑触发的细胞焦亡。然而,其潜在机制仍需进一步阐明。p53在某些条件下抑制细胞焦亡的潜力也值得研究。
2、p53在细胞焦亡受到的调控
已发现多种p53调控因子能够影响p53介导的细胞焦亡。在巨噬细胞中,MUFAs可以结合FABP4并限制赖氨酸去乙酰化酶sirtuin 1的活性,导致p53乙酰化和激活。激活的p53随后通过增加ASC的表达促进炎症小体组装和细胞焦亡。类似地,在LPS D-半乳糖胺诱导的急性肝衰竭小鼠模型中,sirtuin 1通过sirtuin 1 p53 GPX4 GSDMD途径抑制p53诱导的铁死亡和细胞焦亡。该研究还揭示了p53触发的铁死亡与p53触发的细胞焦亡之间的crosstalk。lncRNA MEG3能够稳定p53。在透明细胞肾细胞癌中,MEG3下调导致p53水平降低、p53相关的细胞焦亡受抑制以及细胞增殖增加。p53聚集抑制剂ReACp53能够挽救由MEG3缺失引起的表型,表明触发细胞焦亡可能是治疗p53聚集相关肿瘤的有效方法。值得注意的是,通过激活p53,MEG3还可以促进铁死亡。
在杜氏利什曼原虫感染的THP-1和J774A.1细胞系中,BLIMP-1负责抑制p53表达,减少ROS产生和NLRP3激活,并抑制细胞焦亡。这些结果解释了杜氏利什曼原虫在感染过程中如何避免先天免疫和细胞焦亡,为内脏利什曼病的治疗提供了信息。在小鼠模型中,KIF23的缺失抑制了支气管上皮细胞中的炎症和细胞焦亡,从而缓解了哮喘症状。有趣的是,p53水平随着KIF23的缺失而降低。p53水平的下降是否有助于细胞焦亡的抑制需要进一步研究。最后,KIF23是p53的转录抑制靶标。p53 KIF23负反馈环的病理相关性值得进一步研究。
四、p53 调节的其它非凋亡性细胞死亡
除了铁死亡、坏死性凋亡和细胞焦亡之外,p53 还参与其他 NACD,比如自噬依赖性细胞死亡(autophagy-dependent cell death),内吞性细胞死亡(entotic cell death),PARP-1依赖性细胞死亡(parthanatos)和副凋亡(paraptosis),另外还可能调节泛凋亡,铜死亡(cuproptosis)和双硫死亡(disulfidptosis)。
1、 p53 调节的自噬依赖性细胞死亡(autophagy-dependent cell death)
自噬是一种保守的分解代谢过程,通过降解细胞质物质以供再利用。自噬始于一个孤立的膜结构,逐渐扩展形成吞噬泡(phagophore),最终形成自噬体(autophagosome)。在此过程中,自噬底物被包裹并随后递送至溶酶体进行降解。通常,自噬在应激(特别是营养缺乏)条件下促进细胞存活。然而,失调或过度的自噬可能导致细胞通过自噬依赖性细胞死亡而消亡。

由于p53通过多种机制调控自噬,它也参与了自噬依赖性细胞死亡。当HCT116细胞用SPHK1抑制剂处理时,p53激活自噬依赖性细胞死亡,这一过程通过敲除自噬的两个关键调控因子beclin 1和ATG5得以挽救。p53还通过激活BNIP3L触发的线粒体自噬在结肠癌细胞中诱导自噬依赖性细胞死亡。p53可能通过转录激活TP53INP1在多种细胞类型中促进自噬依赖性细胞死亡。抗肿瘤药物人参皂苷Rh4和白藜芦醇分别激活p53并在结直肠癌细胞和肺腺癌细胞中诱导自噬依赖性细胞死亡。需要强调的是,自噬介导的细胞死亡不一定是 自噬依赖性细胞死亡 :它可能涉及其他细胞死亡模式,如凋亡和铁死亡。因此,当p53调控的自噬导致细胞死亡时,区分发生的是哪种类型的细胞死亡至关重要。
2、p53调控的内吞性细胞死亡(entotic cell death)
Entosis是一个活细胞被非吞噬细胞吞噬形成 细胞内细胞 结构的过程。细胞骨架重排介导的细胞侵袭在entosis中起关键作用。通常(但不总是),内化的细胞将通过entotic细胞死亡而消亡。

在有DNA损伤的有丝分裂上皮细胞中,p53通过激活RND3促进entotic细胞死亡。这一作用消除了非整倍体子细胞以维持基因组完整性。有趣的是,突变体p53也被报道促进entosis的形态特征 细胞内细胞结构 这可能具有促肿瘤作用,因为内化的细胞可能以某种方式逃避细胞死亡。此外,p53驱动的皮质祖细胞entotic细胞死亡与PALS1相关的小头有关。
3、p53调控的PARP-1依赖性细胞死亡(parthanatos)
在轻度DNA损伤后,PARP1被激活以修复DNA并促进细胞存活。然而,当DNA损伤广泛存在时,PARP1被过度激活并产生过量的聚(ADP-核糖)(PAR)聚合物,这些聚合物促进线粒体中AIF的释放。在细胞质中,AIF与MIF结合,AIF MIF复合物转运到细胞核中将基因组DNA切割成大片段,导致parthanatos。

在人类结直肠癌和乳腺癌细胞以及小鼠胚胎成纤维细胞中,p53可以通过促进PARP1的酶活性来响应ROS诱导的DNA损伤,从而激活parthanatos。在CRC中,当激酶AKT被抑制时,p53还通过与PARP1直接相互作用促进PAR的聚合,从而激活parthanatos,这一过程阻碍了CRC细胞的生长。此外,p53可能通过转录激活AIF来促进parthanatos。然而,有报道显示,在存在BRAF V600E突变的情况下,p53缺失使肿瘤细胞对ETS抑制剂诱导的parthanatos更加敏感。因此,p53在parthanatos触发的肿瘤抑制中是被激活还是被抑制,可能取决于具体情况。
4、p53调控的副凋亡(paraptosis)
Paraptosis是一种不依赖于半胱天冬酶的NACD;它可以由许多化学化合物和各种应激(如氧化应激和内质网应激)诱导。paraptosis的分子机制细节尚不明确。paraptosis的关键形态特征包括通常与ER和/或线粒体肿胀相关的细胞质空泡化。
在CRC细胞中,人参皂苷Rh2处理诱导了p53依赖的凋亡和paraptosis,共同实现了抗肿瘤效果。然而,p53诱导的空泡形成和paraptosis的确切机制仍不清楚。
5、p53在调控泛凋亡、铜死亡和双硫死亡中的潜在作用
泛凋亡PANoptosis是一种先天免疫炎症性细胞死亡,涉及细胞焦亡、凋亡和坏死性凋亡之间的crosstalk。因此,它具有这三种细胞死亡模式的特征,但不能单独用其中任何一种来解释。PANoptosis的分子机制围绕多种PANoptosome的形成,这些平台整合了细胞焦亡、凋亡和坏死性凋亡信号。由于p53是这三种细胞死亡模式的有效调控因子,因此可以合理推测p53也可以调控PANoptosis。
铜死亡是一种依赖铜的NACD。细胞内铜水平的异常增加促进三羧酸循环(TCA)中脂酰化蛋白(特别是DLAT)的聚集,导致蛋白毒性应激并最终引发铜死亡。尽管缺乏直接证据,但p53被认为在调控铜死亡中具有重要作用。(注:复旦大学的周祥教授课题组近期在 Mol Cell发文首次证明p53的确可以激活铜死亡)
双硫死亡是一种在葡萄糖饥饿条件下由过量二硫化物触发的NACD,通常发生在高表达SLC7A11的细胞中。p53可能通过抑制SLC7A11表达来增强细胞对二硫化物死亡的抵抗。在p53缺陷的癌细胞中,SLC7A11水平升高可能使细胞对二硫化物死亡更加敏感,这为治疗这些癌症提供了一个潜在的治疗弱点。
五、靶向p53调控的非凋亡性细胞死亡用于疾病治疗
由于p53介导的NACD与多种疾病的病理相关,大量研究致力于开发靶向p53-NACDs的治疗药物。在本节中,我们以p53调控的铁死亡、坏死性凋亡和细胞焦亡为例,展示这一方法在临床应用中具有的巨大潜力。

1、靶向p53调控的铁死亡
癌症是与靶向铁死亡最相关的疾病。许多药物已被发现能够通过激活p53介导的铁死亡来杀死癌细胞。例如,gambogenic acid通过激活p53在上皮-间质转化细胞中触发铁死亡,从而抑制黑色素瘤的转移。抗寄生虫药物氟苯达唑通过激活p53同时诱导铁死亡和细胞周期阻滞,有效抑制去势抵抗性的肿瘤生长。三萜皂苷衍生物D13通过激活p53调控的铁死亡和凋亡,表现出强大的杀伤多药耐药癌细胞的功效。p53激活的铁死亡还可以与其他药物联合用于治疗癌症。PARP抑制剂在治疗保留野生型BRCA1和BRCA2基因的卵巢癌中效果较差。有趣的是,研究发现PARP抑制剂奥拉帕尼通过激活p53在BRCA功能正常的卵巢癌中诱导铁死亡。奥拉帕尼与铁死亡诱导剂FINs联合治疗对这些癌细胞具有协同杀伤作用。双重靶向PI3K和HDAC的抑制剂BEBT-908可以通过乙酰化p53促进铁死亡。重要的是,铁死亡的诱导可能建立促炎性肿瘤微环境,从而增强免疫治疗的效果。然而,病理激活的中性粒细胞中的铁死亡可能负调控抗肿瘤免疫。因此,在临床实践中实现p53激活药物的癌细胞特异性递送至关重要。此外,铁死亡是p53在放疗中抗肿瘤作用的基础。表达突变体p53的癌细胞可能因p53诱导的铁死亡缺失而对放疗产生耐药性,因此可以将FINs与放疗联合用于治疗这些癌症。
除了癌症,通过Nutlin-3激活p53调控的铁死亡有助于阻断疟原虫的肝脏阶段感染。
尽管p53介导的铁死亡在上述情况下对健康有益,但其在正常细胞中的异常诱导可能导致器官损伤或神经退行性疾病(NDD)。在这些情况下,应避免铁死亡。在叶酸诱导的急性肾损伤中,补充 -硫辛酸有效抑制p53介导的铁死亡,从而减轻肾损伤。在小鼠模型中,芍药苷通过直接结合p53并抑制铁死亡,促进认知行为的改善。
需要指出的是,上述大多数通过p53介导铁死亡的药物都是通过p53抑制经典的SLC7A11 GPX4途径发挥作用。然而,正如我们所讨论的,非经典铁死亡途径在p53功能中具有重要作用。因此,也应关注p53调控的这些非经典铁死亡途径及其治疗潜力。
2、靶向p53调控的坏死性凋亡
坏死性凋亡既可能具有肿瘤抑制作用,也可能具有促肿瘤作用。然而,目前的研究表明,p53介导的坏死性凋亡主要具有肿瘤抑制作用。p53激活的坏死性凋亡可能与p53的其他功能协同作用以消除癌细胞。在胶质母细胞瘤中,免疫调节药物FTY720通过ROS JNK p53途径诱导坏死性凋亡,从而抑制肿瘤生长。在胶质瘤中,沙利霉素介导p53向线粒体的转运,p53在线粒体中与CypD相互作用并诱导线粒体通透性转换孔(PTP)相关的坏死性凋亡。
在人类T细胞病毒1型转化的成人T细胞白血病细胞中,DHODH抑制剂BAY2402234通过诱导多种细胞死亡模式(包括凋亡、铁死亡和坏死性凋亡)显示出治疗价值。有趣的是,BAY2402234处理也会激活p53。p53是否参与了BAY2402234触发的坏死性凋亡需要进一步研究。治疗,激活p53可能是一种有前景的方法。此外,诱导坏死性凋亡可能对神经母细胞瘤具有新的治疗效果。因此,研究是否可以通过激活p53调控的坏死性凋亡有效根除神经母细胞瘤细胞将是一个有趣的课题。
与p53激活的铁死亡类似,p53诱导的坏死性凋亡也可能对正常器官造成损伤。例如,p53促进坏死性凋亡,从而加重酒精性肝病(ALD)。姜黄素通过增加NRF2的表达来改善ALD,NRF2抑制p53的活性。CypD诱导的坏死性凋亡与多种脑疾病(如神经退行性疾病)相关。(CsA)和其他配体可以破坏CypD p53复合物。这些抑制线粒体CypD p53相互作用的药物可能有助于治疗由CypD p53 坏死性凋亡轴引起的疾病。
3、靶向p53调控的细胞焦亡
在结直肠癌中,人参皂苷Rh3通过STAT3 p53 NRF2途径同时激活细胞焦亡和铁死亡,从而抑制癌细胞的生长。p53激活的自噬有助于细胞焦亡,可能导致高尿酸血症肾病。自噬抑制剂3-甲基腺嘌呤可以有效保护肾脏免受细胞焦亡的影响,并改善近端肾小管细胞中的高尿酸血症肾病。在大肠杆菌感染后,p53诱导的细胞周期阻滞和凋亡可能导致牛乳腺上皮细胞中的细胞焦亡。靶向p53或细胞焦亡的特异性抑制剂可能具有预防大肠杆菌诱导的牛乳腺炎的潜力。Curaxin CBL0137是一种p53激活药物。CBL0137诱导caspase-3 GSDME依赖的细胞焦亡,从而杀死卵巢癌细胞。这一效应依赖于p53靶基因BAX在线粒体膜上的积累。p53 BAX轴可能是CBL0137治疗引起的细胞焦亡的部分机制。
总之,目前存在多种针对p53介导的非凋亡性细胞死亡(NACDs)的治疗方法,适用于不同的病理条件。未来,希望更多具有这种活性的药物能够造福于患有相关疾病的个体。
六、结论与未来展望
在过去几年中,关于p53调控的铁死亡、坏死性凋亡、细胞焦亡及其他非凋亡性细胞死亡的机制、调控和治疗意义,我们已经获得了许多见解。然而,仍有许多问题需要解决,以下是其中一些主要问题的讨论。
从概念上讲,为什么p53能够调控如此多的细胞死亡途径?
一种可能性是,作为细胞的守护者,p53必须有效清除受损细胞;诱导不同类型的细胞死亡可以确保这一任务的完成。p53在铁死亡中的作用已被深入研究。然而,我们对p53在其他NACDs中的功能机制及其生理或病理相关性的理解仍然较为初步。需要更多努力来阐明这些问题。一个有趣的现象是,许多受p53抑制的靶基因,如SLC7A11、VKORC1L1和PHGDH,对p53调控的NACD有重要贡献。目前研究主要集中在受p53激活的靶基因上,未来应更多关注p53在NACD中抑制的靶标。值得注意的是,p53在NACDs中影响的许多靶标并非p53的直接转录靶基因,而是p53通过间接方式来影响它们的表达。
从机制上讲,一个主要研究任务是识别在特定条件下决定p53激活的特定NACD类型的因子。刺激和其他上游信号在这一过程中起着关键作用。然而,某些刺激(如DNA损伤和ROS)和信号通路(如TNF和TLR通路)可能影响不同的细胞死亡模式。一个相关的问题是,p53对NACDs的某些调控效应表现出细胞类型特异性或组织特异性。不同的应激和信号与其他细胞类型特异性或组织特异性调控因子协同作用,汇聚于p53,以context-dependent的方式促进特定的NACD。例如,在应对DNA损伤或ROS等急性应激时,p53可以同时诱导凋亡和铁死亡以清除应激细胞。在这些应激条件下,哪种细胞死亡途径占主导地位确实是一个复杂的问题,值得进一步研究。p53似乎倾向于在应对氧化应激时诱导铁死亡,如在培养的癌细胞中用TBH处理时,p53调控的铁死亡可能在时间上先于凋亡。除了相对研究较多的凋亡和铁死亡外,p53的某些翻译后修饰(PTMs)或辅因子是否特异性地影响某些类型的细胞死亡?明确p53介导的NACD的调控机制和context依赖性是开发靶向这些途径的治疗药物的前提。
最近的研究表明,不同的细胞死亡途径可以相互作用。鉴于p53调控的细胞死亡途径可能共享共同的上游触发因素或重叠的分子机制,这些途径之间的crosstalk是可能的。进一步探索这一可能性在未来将具有重要意义。
在治疗上,靶向p53 NACD途径不仅补充了现有的临床治疗手段,如癌症治疗中的凋亡诱导药物,还在某些特定情况下具有独特优势。值得注意的是,上述大多数治疗药物针对的是表达未突变p53的癌细胞。然而,这些癌细胞通常采用多种机制来减弱野生型p53的活性,这对实现最佳的p53激活提出了挑战。更重要的是,p53在癌症中经常发生突变或缺失。在这些情况下开发靶向p53 NACD的治疗方法具有重要价值。不幸的是,目前在这些癌细胞中恢复野生型p53活性的策略尚不完全令人满意。联合治疗为克服这些障碍提供了一个有前景的途径。此外,p53 NACD途径仅部分贡献于上述大多数靶向药物的治疗效果。这些药物还影响p53的其他功能,甚至涉及p53非依赖的机制。因此,一个关键目标仍然是特异性靶向p53 NACD轴。
铁死亡、坏死性凋亡、细胞焦亡和PANoptosis通常被认为是免疫原性细胞死亡(ICD)的形式,这一过程中细胞死亡可以刺激免疫反应。近年来,调节免疫已成为p53的一项功能。确定p53调控的ICD是否有助于局部或全身免疫反应至关重要,因为这可能对癌症治疗具有重要影响,包括p53激活与免疫治疗的潜在组合。
如果未来发现p53调控其他NACDs,也不会令人惊讶。要充分理解p53 NACD的联系,并利用p53在这些细胞死亡模式中的力量来治疗疾病,未来还需要大量工作与投入。
版权声明 本网站所有注明“来源:100医药网”或“来源:bioon”的文字、图片和音视频资料,版权均属于100医药网网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:100医药网”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 87%用户都在用100医药网APP 随时阅读、评论、分享交流 请扫描二维码下载->