
遵义医科大学附属医院研究者们揭示了低氧诱导心肌梗死后介导心肌纤维化的关键机制 |
 |
来源:100医药网原创 2023-06-15 22:01
心室重构(VR)是从急性心肌梗死(AMI)到心力衰竭发展过程中的重要环节。成纤维细胞的增殖、迁移和表型转化在介导组织愈合和预防不良VR中起着重要作用。心室重构(VR)是从急性(AMI)到心力衰竭发展过程中的重要环节。成纤维细胞的增殖、迁移和表型转化在介导组织愈合和预防不良VR中起着重要作用。因此,调整急性心肌梗死后心肌纤维化水平对于延缓或逆转VR具有重要意义。

图片来源:
近日,来自遵义医科大学附属医院的研究者们在Theranostics杂志上发表了题为 Hypoxia Induces M2 Macrophages to Express VSIG4 and Mediate Cardiac Fibrosis After Myocardial Infarction 的文章,该研究揭示了低氧诱导心肌梗死后M2巨噬细胞表达VSIG4并介导心肌纤维化。
M2巨噬细胞介导的组织修复在急性心肌梗死(AMI)中起重要作用。此外,VSIG4主要表达在组织驻留细胞和M2巨噬细胞上,对稳态的调节至关重要;然而,它在急性心肌梗死中的作用尚不清楚。
本研究采用VSIG4基因敲除和过继骨髓移植嵌合模型,探讨VSIG4在急性心肌梗死中的功能意义。研究者还通过功能得失实验测定心脏成纤维细胞(CFs)的功能。研究者发现,VSIG4促进急性心肌梗死后瘢痕形成和协调心肌炎性反应,同时也促进转化生长因子- 1和IL-10。
此外,研究者还发现,低氧促进了培养的骨髓M2巨噬细胞VSIG4的表达,最终导致CFs转化为肌成纤维细胞。本研究结果揭示了VSIG4在小鼠急性心肌梗死过程中的重要作用,并为急性心肌梗死后纤维化的修复提供了一条潜在的免疫调节治疗途径。
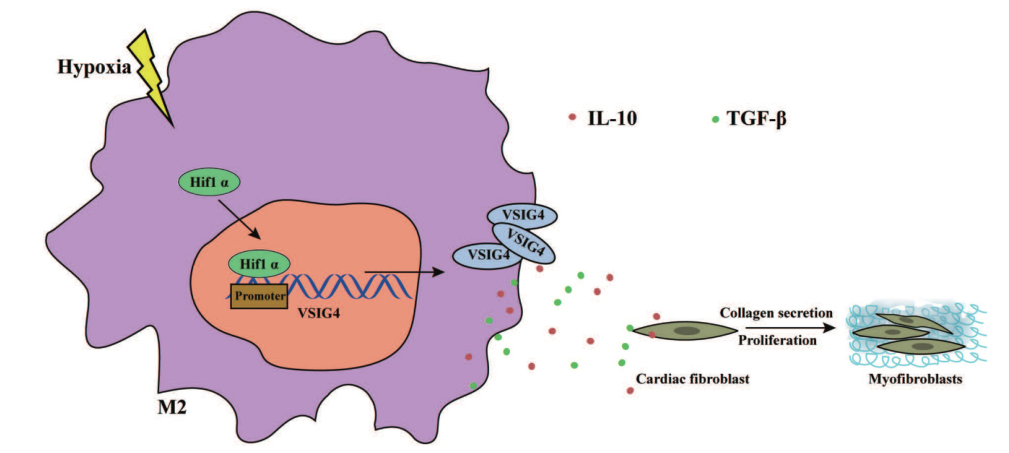
机制总结图
图片来源:
综上所述,研究者证明VSIG4参与了急性心肌梗死后瘢痕的形成。VSIG4协调急性心肌梗死后的心肌炎症反应,促进转化生长因子- -1和IL-10的表达。VSIG4通过间接作用于梗死区的心脏成纤维细胞,加强心脏修复,降低心脏破裂和死亡率。
在心肌梗死的复杂环境中,各种细胞因子、趋化因子和生长因子表达的时空调节可能会影响巨噬细胞。研究者推测,VSIG4+M2巨噬细胞在调节心肌梗死后纤维化修复和功能中的作用,至少部分是由低氧介导的。
进一步探讨缺氧VSIG4+M2巨噬细胞在急性心肌梗死后心脏修复中的作用,确定其在修复过程中的分子水平和功能作用,包括对急性心肌梗死后新生的影响,有待进一步研究。( 100yiyao.com)
参考文献
版权声明 本网站所有注明“来源:100医药网”或“来源:bioon”的文字、图片和音视频资料,版权均属于100医药网网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:100医药网”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 87%用户都在用100医药网APP 随时阅读、评论、分享交流 请扫描二维码下载->
- 相关报道
-
- 2025年药物警戒任务会议召开 (2025-10-24)
- Nat Commun:皮肤细胞“变身”卵子?科学家开创不孕治疗新路径 (2025-10-24)
- 3D微纳机器人研究获进展 (2025-10-24)
- Science:1+1>2的智慧!蚂蚁的“建筑免疫”如何与个体隔离行为协同增强群体抗性 (2025-10-24)
- 西藏社保系统完成跨越式倒退 织密高原“平易近生保证网” (2025-10-23)
- 重磅!杭州医保新政宣布!来岁1月1日起施行 (2025-10-23)
- “搞笑诺贝尔奖”研究,再登Cell子刊:人类也可以通过肛门呼吸,关键时刻能救命,已开展人体临床试验 (2025-10-23)
- 反复腹痛腹泻、家族里多人中招?新研究找到溃疡性结肠炎关键基因 OTUD3,从根源修复肠道屏障有戏了! (2025-10-22)
- Cell Rep:肠道菌群竟会“自制快乐素”?两种益生菌联手改善肠易激综合征 (2025-10-22)
- 招聘启事-浙大医学院附属第一医院尹茂鲁课题组 (2025-10-22)
- 视频新闻
-
- 图片新闻
-









医药网免责声明:
- 本公司对医药网上刊登之所有信息不声明或保证其内容之正确性或可靠性;您于此接受并承认信赖任何信息所生之风险应自行承担。本公司,有权但无此义务,改善或更正所刊登信息任何部分之错误或疏失。
- 凡本网注明"来源:XXX(非医药网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。本网转载其他媒体之稿件,意在为公众提供免费服务。如稿件版权单位或个人不想在本网发布,可与本网联系,本网视情况可立即将其撤除。联系QQ:896150040









