商业广告QQ
896000434
896000434
50条负面清单发布
近日,山东省药品监督管理局发布《医疗器械经营企业、使用单位质量安全主体责任清单和负面清单的通知》(以下简称《通知》)。
《通知》表示,根据《医疗器械监督管理条例》 《医疗器械经营监督管理办法》 《医疗器械使用质量监督管理办法》,山东美国食品药品监督管理局研究制定了医疗器械企业和用户主体责任清单和负面清单。
如下所示:
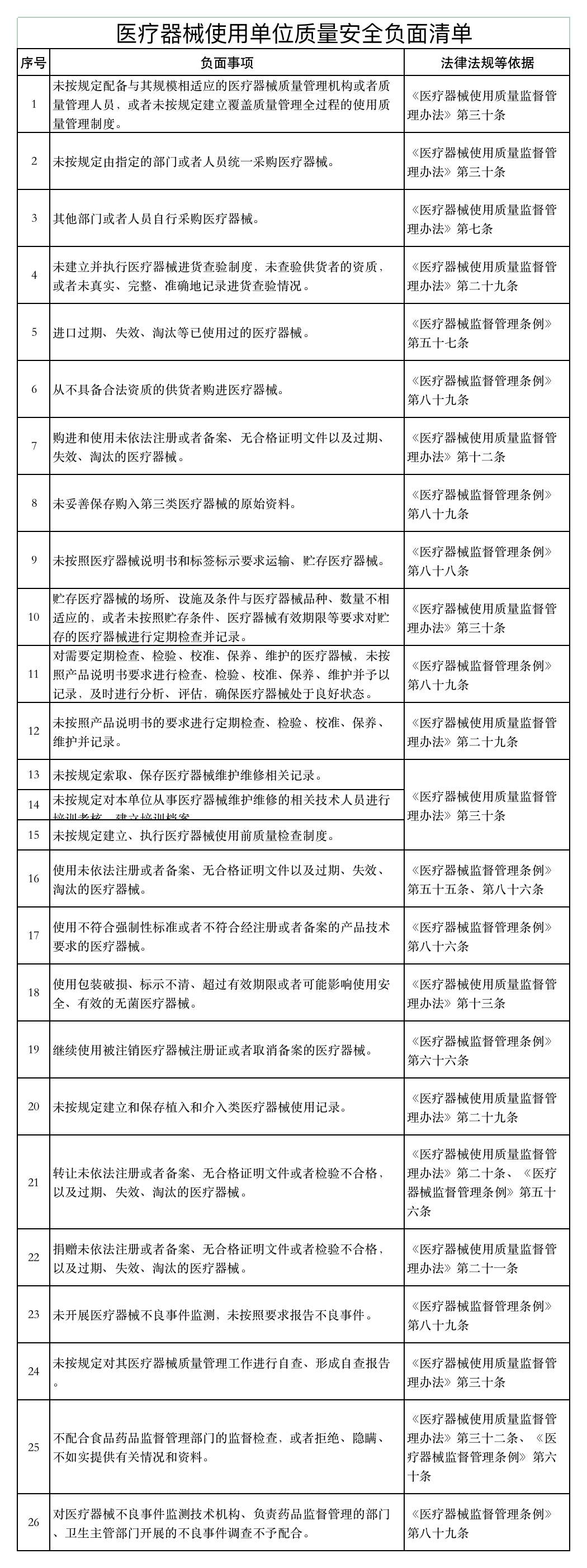
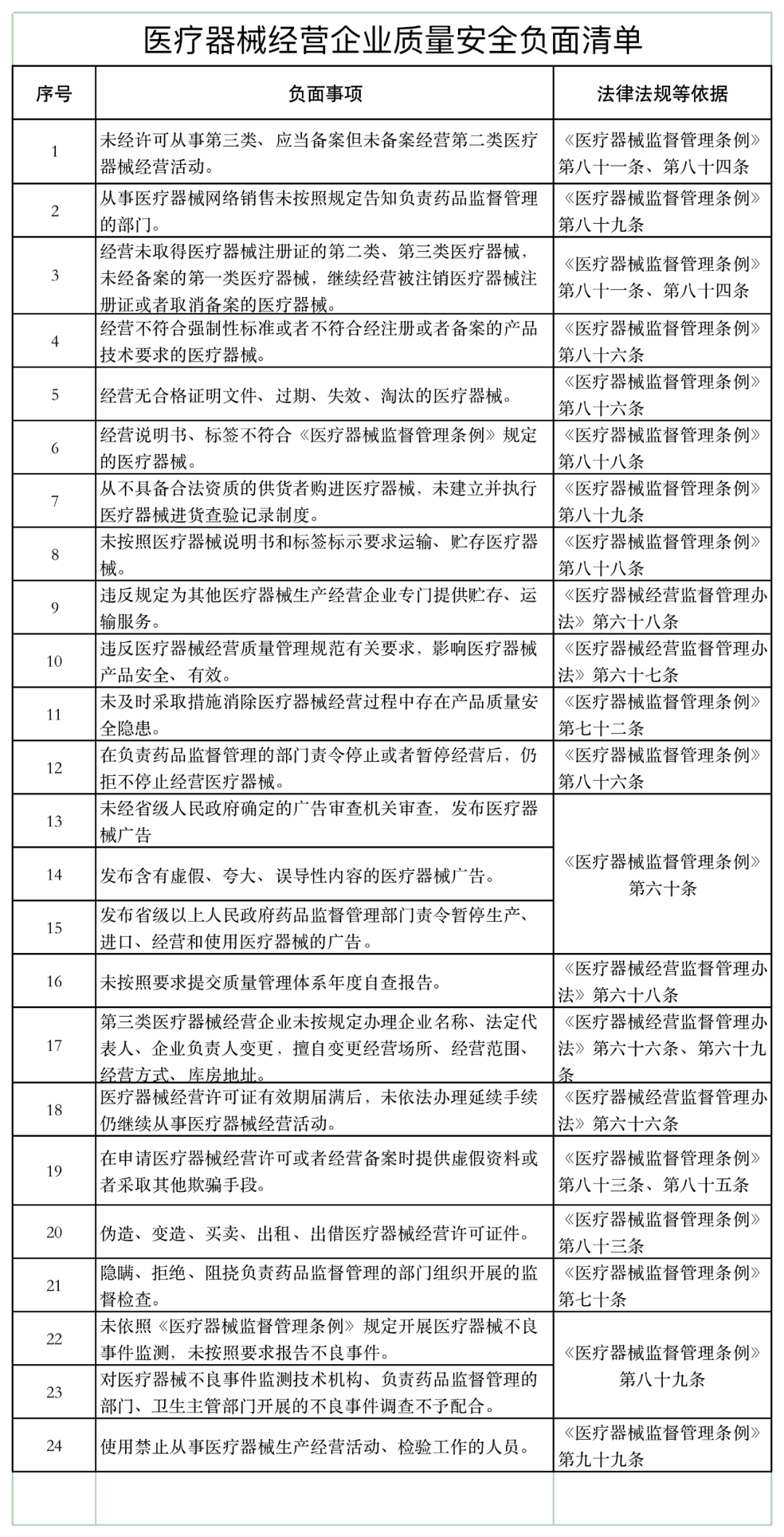
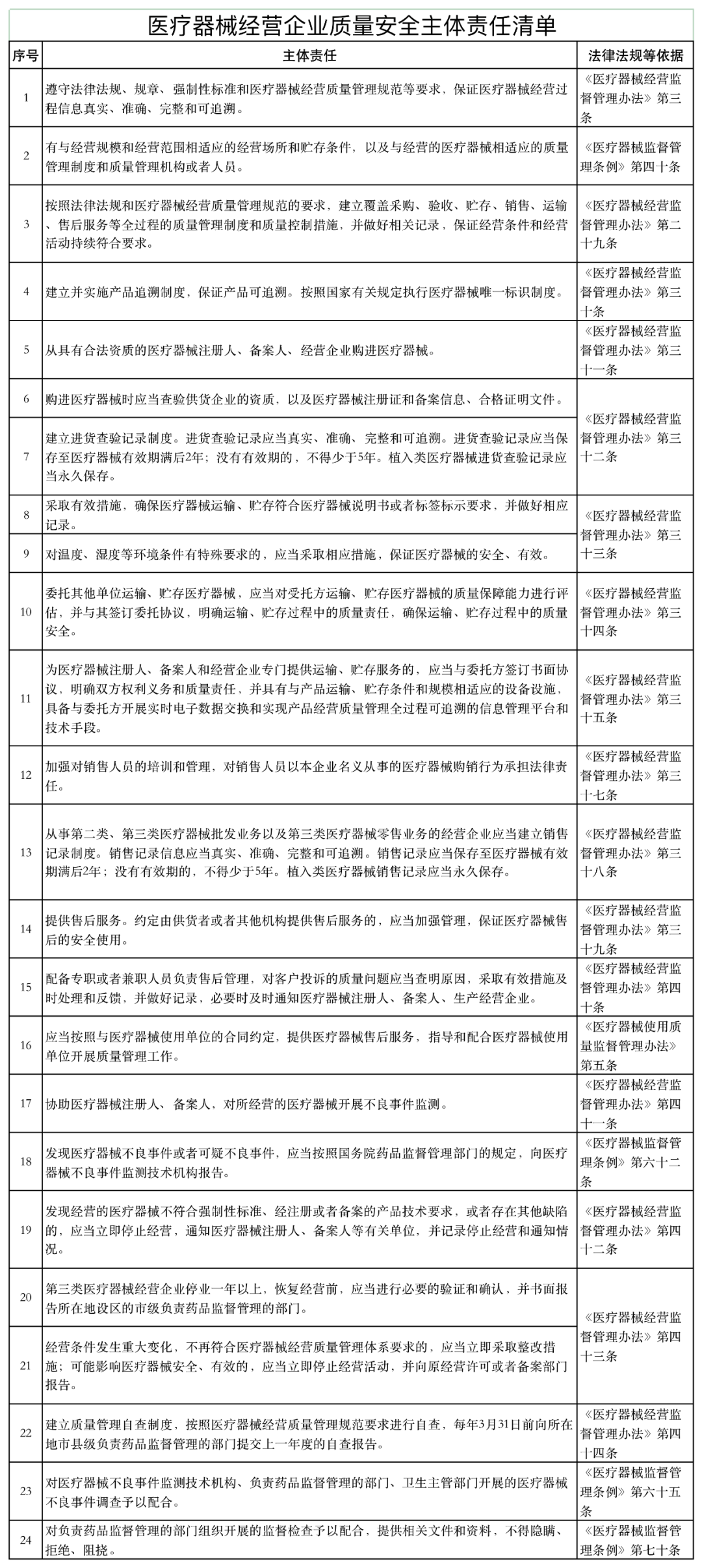
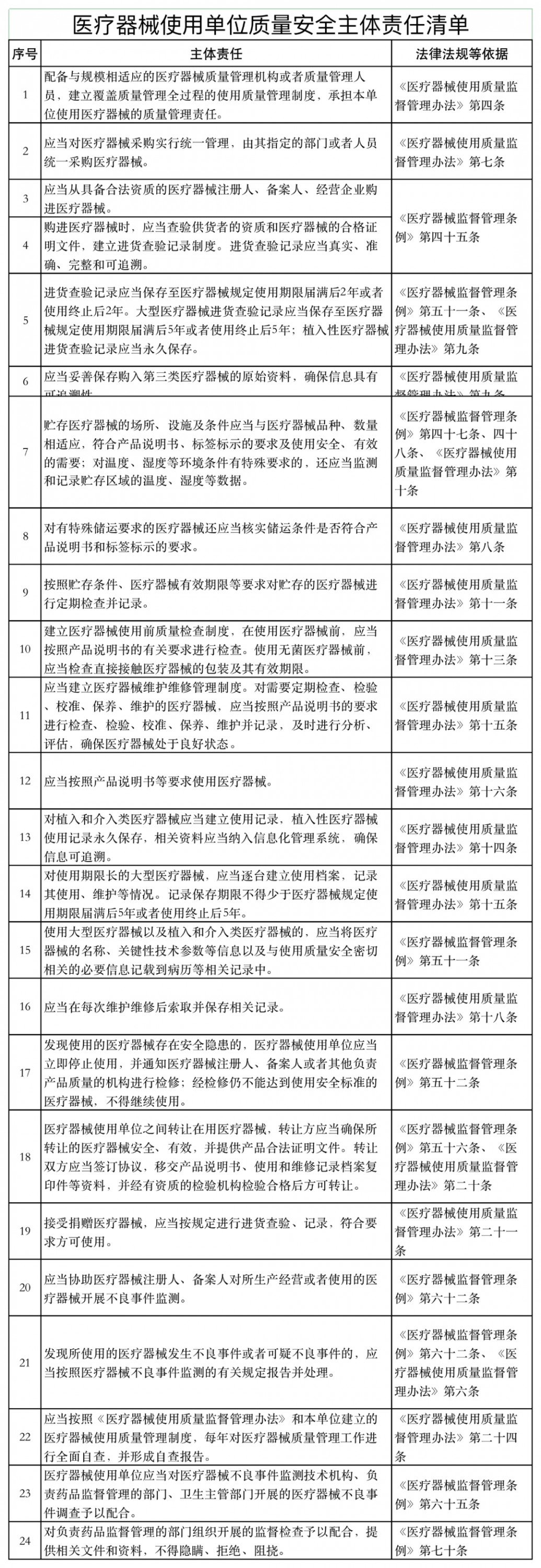
医疗器械经营监管大调整
随着新版《医疗器械生产监督管理办法》 《医疗器械经营监督管理办法》的实施,医疗器械企业的管理和监督被提升到了一个新的纬度。
国家指出,两个办法严格落实“四个最严”要求。落实《医疗器械监督管理条例》条例,全面推行医疗器械注册人备案制度,优化行政许可流程,强化监督检查措施,完善监督检查手段,强化企业主体责任,进一步加大对违法行为的处罚力度。
一方面,03010进一步强化了企业的质量责任。要求企业从事医疗器械经营的,应当按照法律法规和医疗器械经营质量管理规范的要求,建立覆盖采购、验收、储存、销售、运输和售后服务全过程的质量管理体系和质量控制措施,并做好相关记录,确保经营条件和经营活动持续符合要求。
同时,医疗器械经营企业应当建立质量管理自查制度,按照医疗器械经营质量管理规范的要求进行自查,并于每年3月31日前将上一年度的自查报告报送所在地市县级负责药品监督管理的部门。
另一方面,更注重运营全过程的质量管理。
一是要求医疗器械在经营企业时从具有合法资质的医疗器械注册人、备案人和经营企业处采购医疗器械,严格控制购销环节的资质审查,确保医疗器械在合法的流通
二是突出了进货检验和销售的记录要求,保证了产品的可追溯性,应当按照国家有关规定实施医疗器械唯一标识制度。
再次,强调低温和冷藏医疗器械的储存和运输要求,保证产品的运输质量。第四,对运营企业的售后服务提出要求,保证产品的安全使用。
新《经营办法》通过以下措施加强监管:一是实施分类管理。药品监督管理部门应当根据医疗器械企业质量管理和医疗器械产品风险程度,实施分类管理和动态调整。
二是制定年检计划。设区的市、县级药品监督管理部门应当制定年度检查计划,明确监督重点、检查频次和覆盖范围,并组织实施。三是进行延伸检查。
药品监督管理部门可以根据预防和控制医疗器械质量安全风险的需要,对为医疗器械经营活动提供产品或者服务的其他有关单位和个人进行延伸检查。第四,风险咨询和判断。药品监督管理部门应当根据监督检查、产品抽样、不良事件监测、投诉举报、行政处罚等情况,定期进行风险咨询和判断,做好医疗器械质量安全隐患的排查和防控工作。
信用档案建设。市药品监督管理部门应当建立并更新辖区内医疗器械企业信用档案。
103010年,在医疗器械生产许可和备案、监督检查、责任约谈等现有监管方式的基础上,进一步
三是细化明确信息披露和责任约谈制度。药品监督管理部门应当及时公开医疗器械生产许可、备案、监督检查、行政处罚等信息。依法公开,便于公众查询,接受社会监督。医疗器械注册人、备案人和受委托生产企业未采取有效措施消除存在的医疗器械质量安全风险的,药品监督管理部门可以对医疗器械注册人、备案人和受委托生产企业的法定代表人或者负责人进行责任约谈。
委托生产如何开展?
医疗器械注册人和备案人委托生产的,应当按照国家美国食品药品监督管理局制定的《委托生产质量协议指引》的要求,对受托人的质量保证能力和风险管理能力进行评估,与其签订质量协议和委托协议,并监督受托人履行相关协议约定的义务;
受委托生产企业应当按照法律、法规、规章、医疗器械生产质量管理规范、强制性标准、产品技术要求、委托生产质量协议等要求组织生产,对生产行为负责,接受医疗器械注册人和备案人的监督。
委托生产企业应当向原生产许可证或者生产备案部门申报增加的产品品种,并提供委托方、委托产品、委托期限等信息;
增加生产产品涉及生产条件变化,可能影响产品安全性和有效性的,应当在增加生产产品前30个工作日向原生产许可部门报告,原生产许可部门应当及时进行现场核查。许可事项发生变化的,按规定办理相关许可变更。
医疗器械注册人和备案人负责产品上市放行,建立产品上市放行规则,明确放行标准和条件,审核医疗器械生产过程记录和质量检验结果。符合标准和条件的,由授权放行人员签字后方可上市。
委托生产的,医疗器械注册人和备案人还应当审查委托生产企业的生产放行文件。
受委托生产企业应当建立生产放行制度,明确生产放行的标准和条件,确认符合标准和条件后方可出厂。不符合合法法律、法规、规章和强制性标准以及注册或备案产品技术要求的,不得出厂和上市。医疗器械注册人和备案人不得委托受委托的生产企业上市放行。
实行产品品种报告制度。医疗器械生产企业应当向药品监督管理部门申报生产的产品品种。
增加产品品种的,应当向原生产许可或者生产备案部门申报。涉及委托生产的,还应当提供委托方、委托产品、委托期限等信息。增加产品产量的医疗器械生产企业涉及生产条件发生变化可能影响产品安全性和有效性的,应当在增加产品产量前30个工作日向原生产许可部门报告,原生产许可部门应当及时进行现场核查。许可事项发生变化的,按规定办理相关许可变更。
