商业广告QQ
896000434
896000434

OPA1是一种线粒体动力蛋白样GTP酶,负责线粒体形态发生、融合和能量动力学。鉴于OPA1在控制线粒体结构和能量方面的作用,作者推测OPA1可能是通过线粒体依赖机制来调节铁死亡的关键节点。为了研究OPA1在铁死亡中的作用,作者使用了小鼠胚胎成纤维细胞(MEFs)和人类U2OS细胞,并在这两种细胞中分别敲除Opa1,通过三种方法(半胱氨酸剥夺、小分子抑制剂诱导和脂质过氧化检测)诱导铁死亡。研究发现,OPA1的缺失可以显著减少脂质过氧化和由多种铁死亡诱导剂引起的细胞死亡。这表明OPA1可能是铁死亡的一个正调控因子,在铁死亡中起到促进作用。此外,OPA1的GTPase活性在这个过程中十分重要,GTPase功能缺陷突变体(K301A)不能拯救Opa1缺失导致的铁死亡抑制。
研究人员进一步的机制研究发现,OPA1促进铁死亡的能力与其促进线粒体融合的能力无关,因为在缺失Opa1的细胞中表达L-OPA1(具有促进线粒体融合能力)和S-OPA1(不具有促进线粒体融合能力)两种异构体时,所有细胞的死亡率相似,脂质过氧化水平也无显著差异。除了介导线粒体融合外,OPA1还调控线粒体嵴的超微结构、电子传递链复合物(ETC)的组装以及氧化磷酸化。因此,缺失Opa1导致线粒体嵴的结构混乱和ETC I、III、IV和V的组装受损,但不影响复合物II的组装。有趣的是,OPA1在通过半胱氨酸剥夺和xGT抑制剂(erastin)导致的线粒体ROS和脂质ROS积累中发挥作用,而在通过GPx4抑制剂(RSL3)引发的ROS积累中不发挥作用。这种差异性解释了为什么Opa1缺失的MEFs对erastin诱导的铁死亡比对RSL3更具抗性。
为了理解OPA1缺失如何赋予细胞对铁死亡的抗性,研究人员接下来探究了OPA1缺失是否影响xCT-GSH-GPx4通路。研究发现,OPA1的缺失上调了xCT基因的转录和活性,进而GSH水平也得到提升。更加重要的是,OPA1缺失细胞表现出更高的GPx4蛋白水平,并在erastin和RSL3处理期间GPx4水平并没有很快的下降,这与细胞对铁死亡的抗性一致。敲低xCT基因使得OPA1缺失细胞对铁死亡的抗性消失,细胞重新变得对铁死亡敏感。这些结果表明,OPA1缺失通过上调xCT-GSH-GPx4通路赋予细胞对铁死亡的抗性。研究人员随后通过RNA-seq分析发现,ATF4转录因子是整合应激反应的关键转录激活因子,能够上调一些铁死亡抗性相关基因(如xCT基因)的表达,OPA1的缺失导致ATF4表达上调以及整合应激反应的激活。
文章的最后,作者还探究了OPA1对脂质代谢的影响。通过脂质组学分析,磷脂、溶血磷脂、甘油酯、游离脂肪酸和鞘脂在OPA1缺失的细胞中表现出显著变化。其中,多不饱和脂肪酸的某些种类表现出减少的现象,大多数单不饱和脂肪酸则显著富集。多不饱和脂肪酸在铁死亡过程中特别容易发生过氧化,而单不饱和脂肪酸可以减少细胞膜上脂质过氧化的积累,这表明单不饱和脂肪酸的积累可能在抑制铁死亡中发挥了潜在作用。
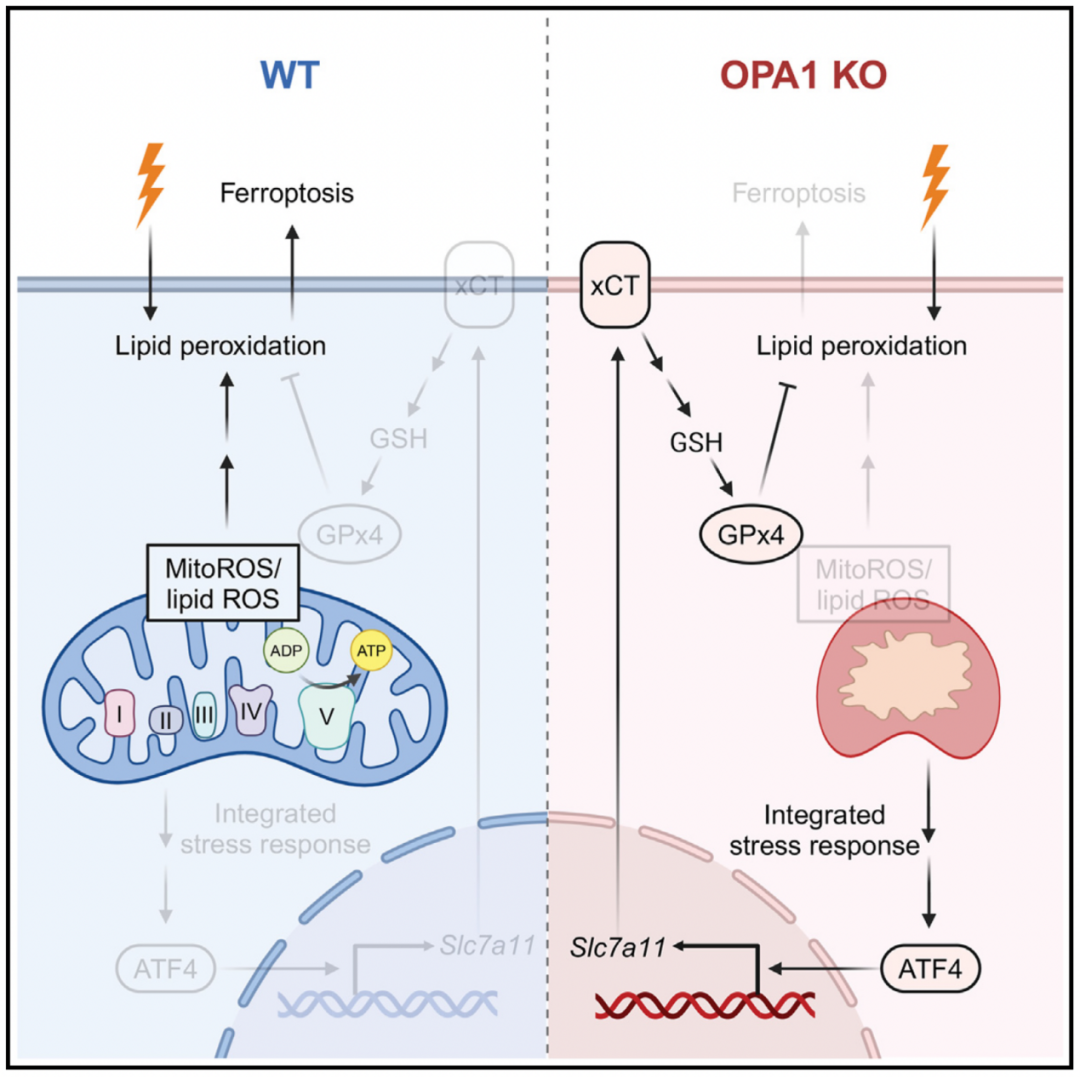
模式图(Credit:Molecular Cell)
