商业广告QQ
896000434
896000434

合成致死互作(Synthetic lethal interactions)是指当两个基因中的任意一个发生突变时,细胞仍然能够存活,但当这两个基因同时发生突变时,细胞就会死亡【3】。由于酵母基因之间的合成致死互作增加了它们的人类直系同源基因之间存在类似相互作用的可能性,作者因此首先鉴定了酵母中与核心自噬基因有至少5个负向遗传或合成致死互作的人类直系同源基因。通过对人类同源基因的CRISPR/Cas9敲除实验,作者鉴定到与自噬缺陷细胞(如ATG16L1-null和ATG9-null细胞)相关的潜在合成致死互作,尤其是在蛋白酶体和核孔复合体成分(如PSMD7、NUP98和NUP133)中尤为显著。通过实验手段进一步验证这些互作后,作者确认了蛋白酶体和核孔复合体在维持自噬缺陷细胞存活过程中不可或缺。这些发现表明,核孔蛋白和蛋白酶体活性在自噬缺失细胞中的相互作用在从酵母到哺乳动物细胞间的进化过程中十分保守。
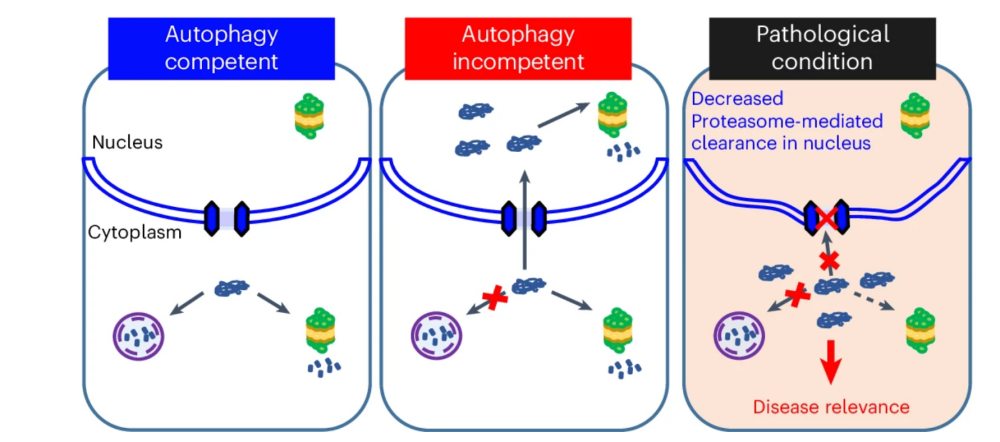
发生突变的A53T -突触核蛋白(A53T -Syn)会通过自噬和蛋白酶体这两条主要的细胞内清除途径进行清除,其清除异常会导致常染色体显性遗传性PD的发生【4】。利用A53T -Syn作为模型底物,作者发现在自噬受损和蛋白酶体抑制条件下,细胞质中的A53T -Syn水平降低,但会在细胞核内过度积累。核孔复合体(NPC)成分及蛋白酶体活性会影响A53T -Syn的细胞核转运。此外,A53T -Syn的入核依赖于核转运蛋白importin ,抑制importin / 运输途径降低了A53T -Syn的核/质比。因此,蛋白酶体活性降低和NPC功能障碍潜在地通过共同分子途径导致自噬缺失细胞的合成适应性丧失。
接下来,作者评估了自噬缺失细胞中新合成蛋白向细胞核的转运是否也发生了激活。通过分析新合成的错误折叠的蛋白质/多肽的亚细胞定位,作者发现抑制自噬会引发大量新合成和错误折叠的蛋白质从细胞质转移到细胞核,进而由核蛋白酶体降解。急性自噬抑制会促进 -Syn A53T与importin 的互作,即自噬底物在自噬受损时与importins的结合增强。可见这一核转运过程与核转运蛋白importin 相关,且受自噬状态的影响。恢复自噬后,蛋白质在核质之间的积累比例也会恢复正常。可见,自噬过程与蛋白质核转运之间存在协同作用,自噬受损会导致细胞质蛋白(可能为自噬底物)涌入细胞核进行核内蛋白酶体降解。
亨廷顿病(HD)是一种常染色体显性神经退行性疾病,与轻微的自噬抑制和核孔复合体(NPC)及核质转运的紊乱有关。作者检测了携带HD突变(HTT125Q)的细胞是否会在应对自噬障碍时,诱导细胞质底物转运至细胞核进行蛋白酶体降解。结果表明,HD突变细胞的自噬功能异常,并且细胞无法有效地通过将细胞质中的蛋白质转运到细胞核中进行降解来应对自噬障碍下的蛋白质积累。尤其是在使用蛋白酶体抑制剂时,HD突变的iPS细胞衍生神经元、HD患者的成纤维细胞及小鼠纹状体细胞表现出较低的胞质到核的蛋白质转运能力。与正常对照细胞相比,HD突变细胞中新合成蛋白质的核内积累明显减少。因此,HD突变细胞的核质转运机制效率低下,无法有效将自噬底物转运到细胞核中,这导致蛋白质聚集在细胞质中,特别是在神经元中,聚集物多见于细胞体和神经突起。
此外,在HD细胞中,蛋白质聚集物主要集中在细胞质中,无论是抑制自噬还是抑制蛋白酶体功能,都会加剧这种情况。自噬抑制导致更多聚集物在细胞质中积累,而蛋白酶体抑制则增加了细胞质中自噬聚集物的范围和数量。HD细胞不仅有自噬抑制的先天性缺陷,还因核质转运活性不足而更容易由于蛋白质积累导致细胞死亡,这两种缺陷协同作用,加重了细胞的损伤和疾病的进展。
模式图(Credit:Nature Cell Biology)
综上所述,本研究发现蛋白酶体和核孔复合体成分的缺失会导致自噬缺陷细胞出现类似合成适应性丧失的协同生存变化,其原因在于自噬缺陷期间蛋白质会需要从细胞质转运到细胞核中,并在核蛋白酶体中发生降解。此外,由于亨廷顿病等神经退行性疾病发生过程中自噬与核质转运均有缺陷,因此这些细胞在蛋白质稳态紊乱时更易受到损害。
